
Abstract
BACKGROUND:
Clinically non-functioning Pituitary Adenomas (NFPA) are among the most common neoplasms of the sellar region. They usually present with compressive symptoms such as headache and visual field defects and not infrequently, are found incidentally. NFPA are classified as gonadotropinomas, null cell adenomas, according to their immunohistochemical phenotype. The molecular alterations responsible for the development of these lesions are incompletely understood, and there is scarce information regarding the molecular alterations and markers.
OBJECTIVE:
We carried out an in-silico analysis aimed at identifying the molecular alterations in NFPA and to discover new molecular markers.
METHODS:
Twenty-three microarray libraries were analyzed. Fourteen correspond to NFPA and 9 to control tissue gland. They were analyzed using Partek Genomic Suite to identify differentially expressed genes and WebGestalt and Metascape to understand the meaning behind the gene lists.
RESULTS:
Pituitary adenomas showed a markedly different transcriptome compared to the non-tumoral gland, regardless of their putative immunophenotype. Genes related to calcium metabolism such as CACNA2D4, immune-related CXCR4, and stem cell-related KLF8 and PITX2 were altered.
CONCLUSIONS:
Differentially expressed calcium metabolism and immune-related genes in NFPA represent attractive molecular markers and potential therapeutic targets.
Introduction
Pituitary adenomas (PA) are monoclonal tumors arising in adenohypophyseal cells and represent
NFPA are the second most common lesion of the gland, only overcome by prolactinomas. More than 45% of the NFPA show positive immunostaining for
To date, there is scarce information regarding the molecular pathogenesis of sporadic pituitary adenomas in general and of NFPA in particular. Furthermore, little is known about the molecular differences and similarities between the various types of PA. Therefore, in the present work we analyzed the molecular alterations in coding mRNA and non-coding RNA expression and the potential cellular pathways altered in PA using the available microarray data to identify potentially novel molecular diagnostic markers.
Materials and methods
We analyzed data downloaded from the European Bioinformatics Institute (EMBL-EBI) and from Gene Expression Omnibus (GEO). The RNA libraries were selected according to the following criteria: a) The tissue of origin had to be a corroborated pituitary adenoma obtained at surgery; b) The RNA had to be analyzed using the same microarray; and c) The data had to meet quality control parameters such as Pearson and Spearman indices, as well as labeling and hybridization constraints.
Transcriptome in silico analysis
For transcriptome analysis, a total of 14 PA samples and 9 non-tumoral control glands were selected for coding mRNA and non-coding RNA. These data correspond to GSE26966. Dataset analyses were achieved by means of CEL files with the Expression Console, Partek Genomics Suite 6.6v software (Partek Incorporated, Saint Louis, MO, USA) and Transcriptome Analysis Console (Affymetrix, Santa Clara, CA, USA). Pearson and Spearman’s correlation was performed, and probe sets were summarized by means of Median polish and normalized by quantiles with no probe sets excluded from the analysis. Background noise correction was achieved by means of Robust Multi-chip Average (RMA) and data were log2-transformed. Data grouping and categorization was achieved by principal component analysis (PCA). Differentially expressed genes were detected by means of ANOVA. Genes were considered altered with
Pathway, enrichment networks, protein-protein interactions
WebGestalt (
Results
Molecular alterations in gonadotrope cell adenomas, altered transcripts and cellular pathways of the pituitary lesions
Our main interest was to perform whole transcriptome analysis to identify molecular alterations in GNFPA in an attempt to elucidate potential new therapeutic targets. Interestingly, at first glance, the PCA showed a readily distinctive transcriptome between control pituitary and tumor samples (Fig. 1). The results showed 2382 altered genes, 1024 upregulated and 358 genes down regulated, showing good discrimination between normal and pituitary adenomas (Fig. 2).
The principal component analysis shows the formation of two groups, normal gland (blue dots), and NFPA (red dots) indicating different transcriptomes.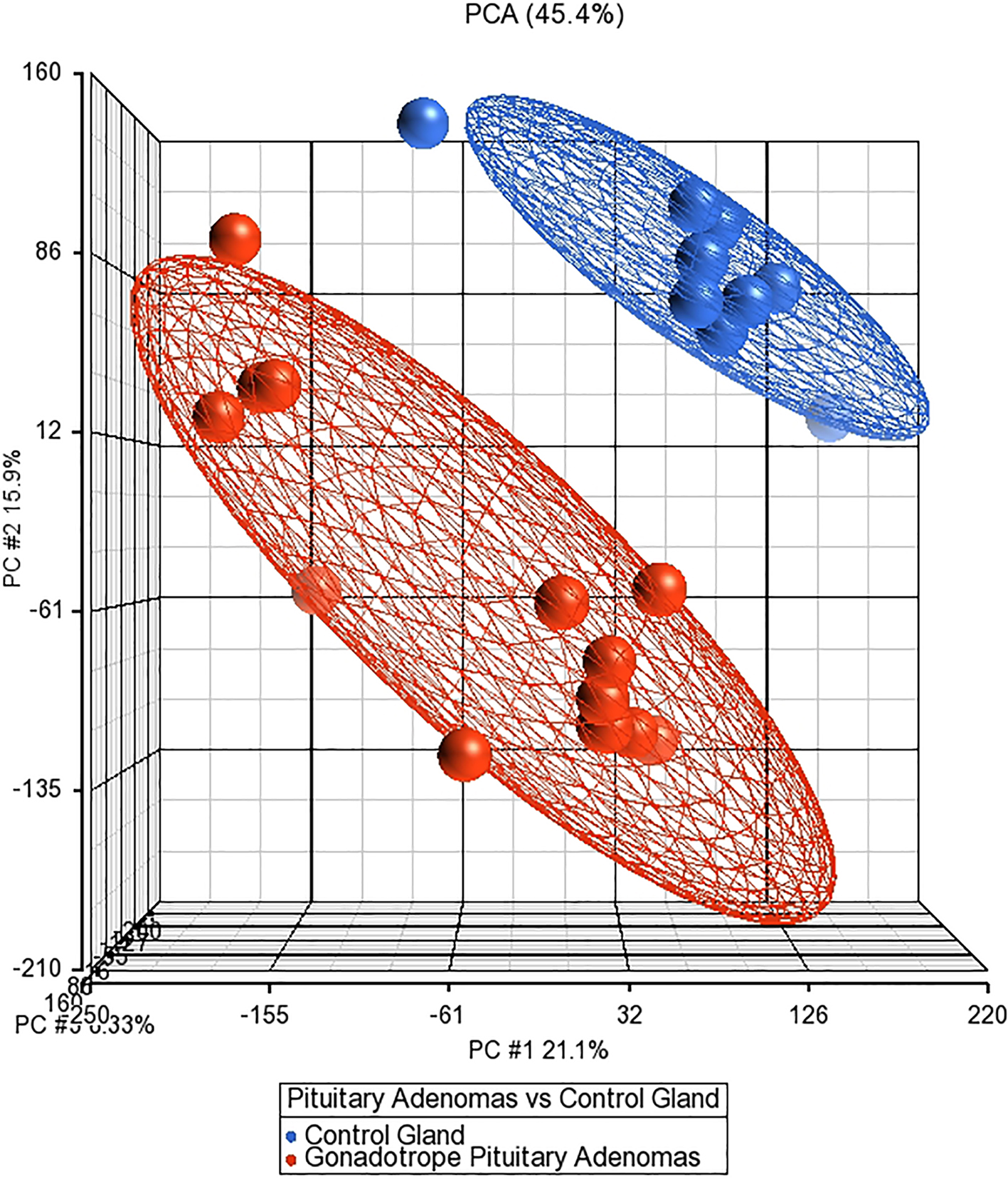
a. Corresponds to whole transcriptome heat map from NFPA, and Normal pituitary gland, showing molecular dissimilarities between NFPA and normal gland. b. Depicts the heat map corresponding to the differentially expressed genes in NFPA types compared to the normal pituitary gland.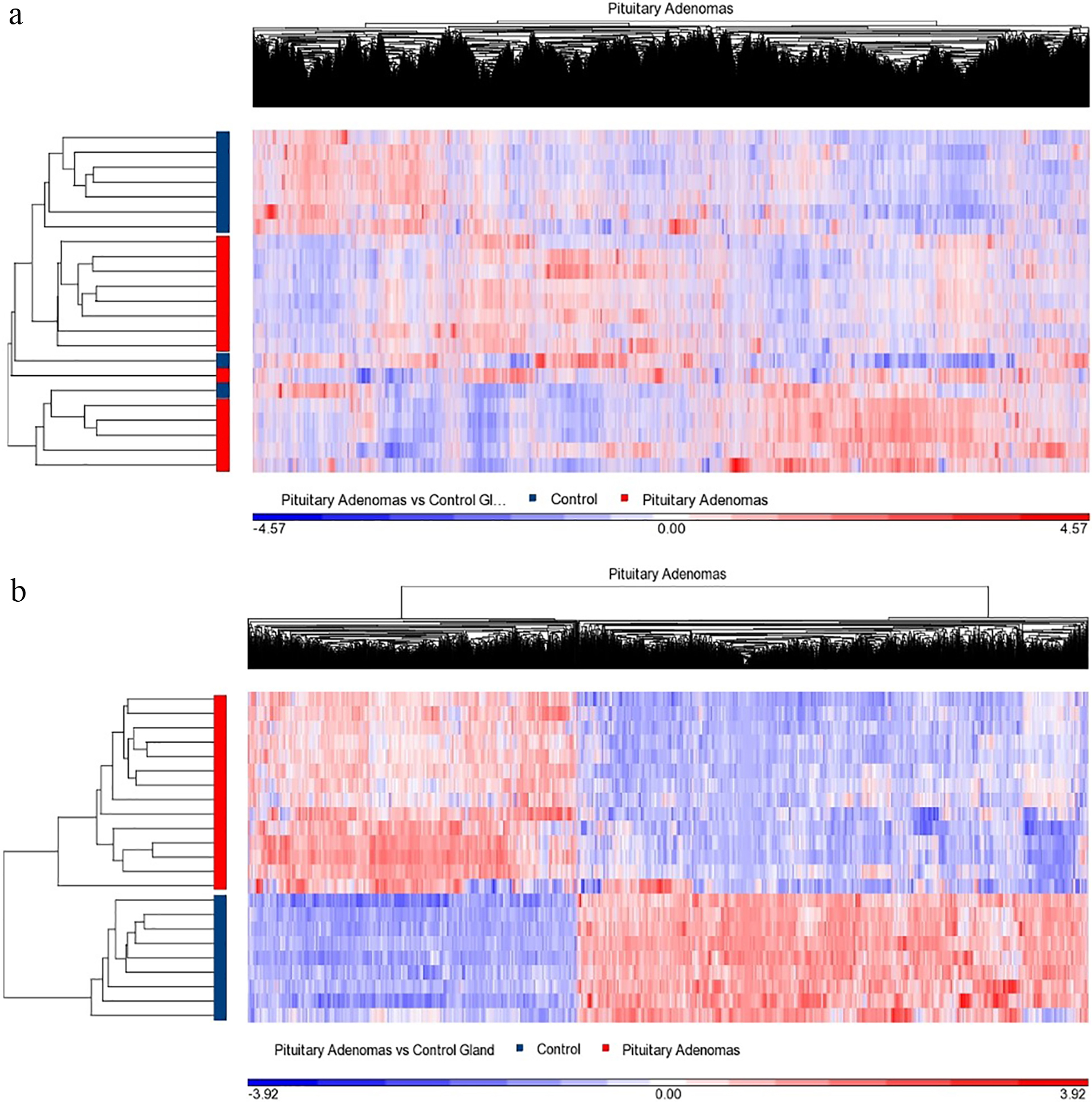
Dot blots from representative altered expression genes. a. Represents CACNA2D4 upregulated gene expression in NFPA compared to the normal pituitary gland. b and c. Portray PITX2 and LINC01616, respectively, gene up-regulation in both pituitary lesions. d–f. Shows CXCR4, STAT3 and LINC00632 genes down-regulated in both pituitary lesions compared with the normal gland. Blue dots represent control samples and red dots represent NFPA.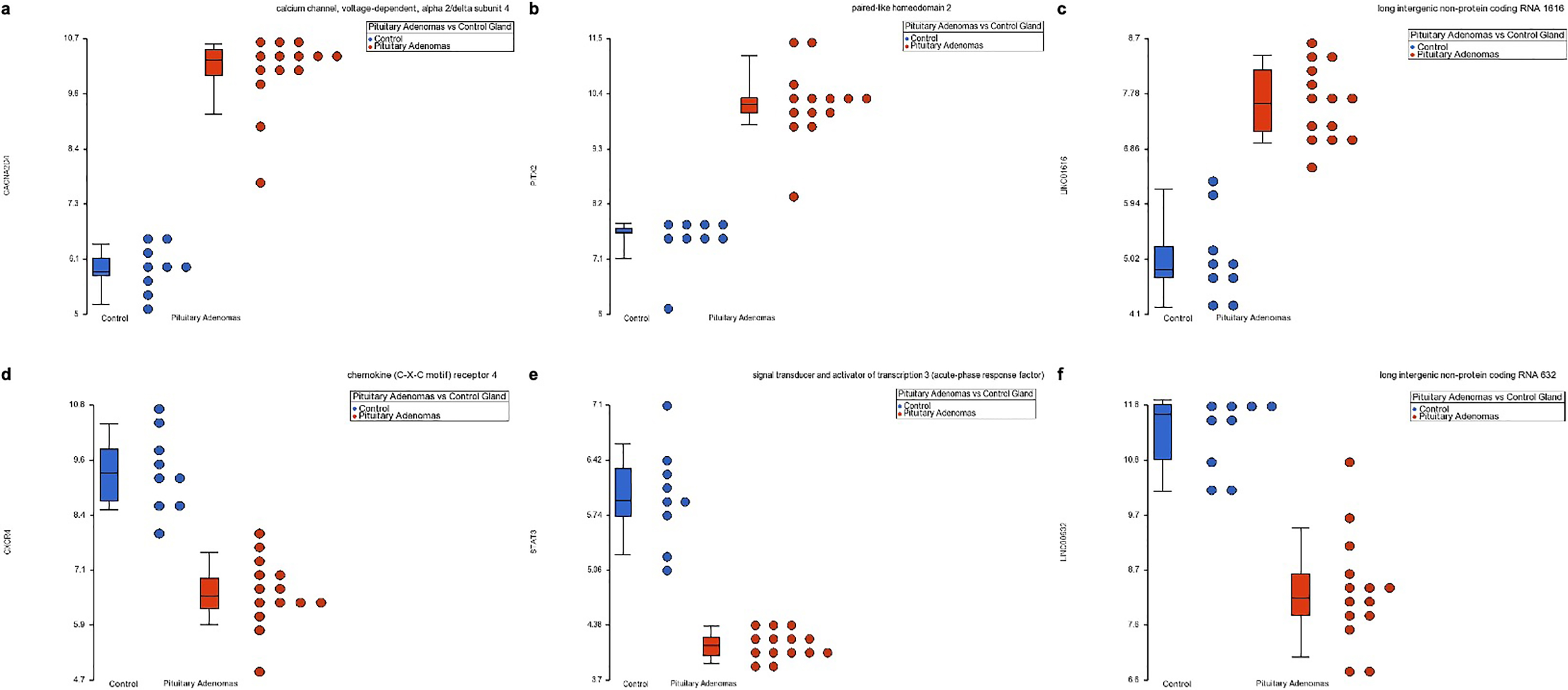
a. Shows the pathways in which upregulated genes are involved. b. Shows the pathways in which down-regulated genes are involved.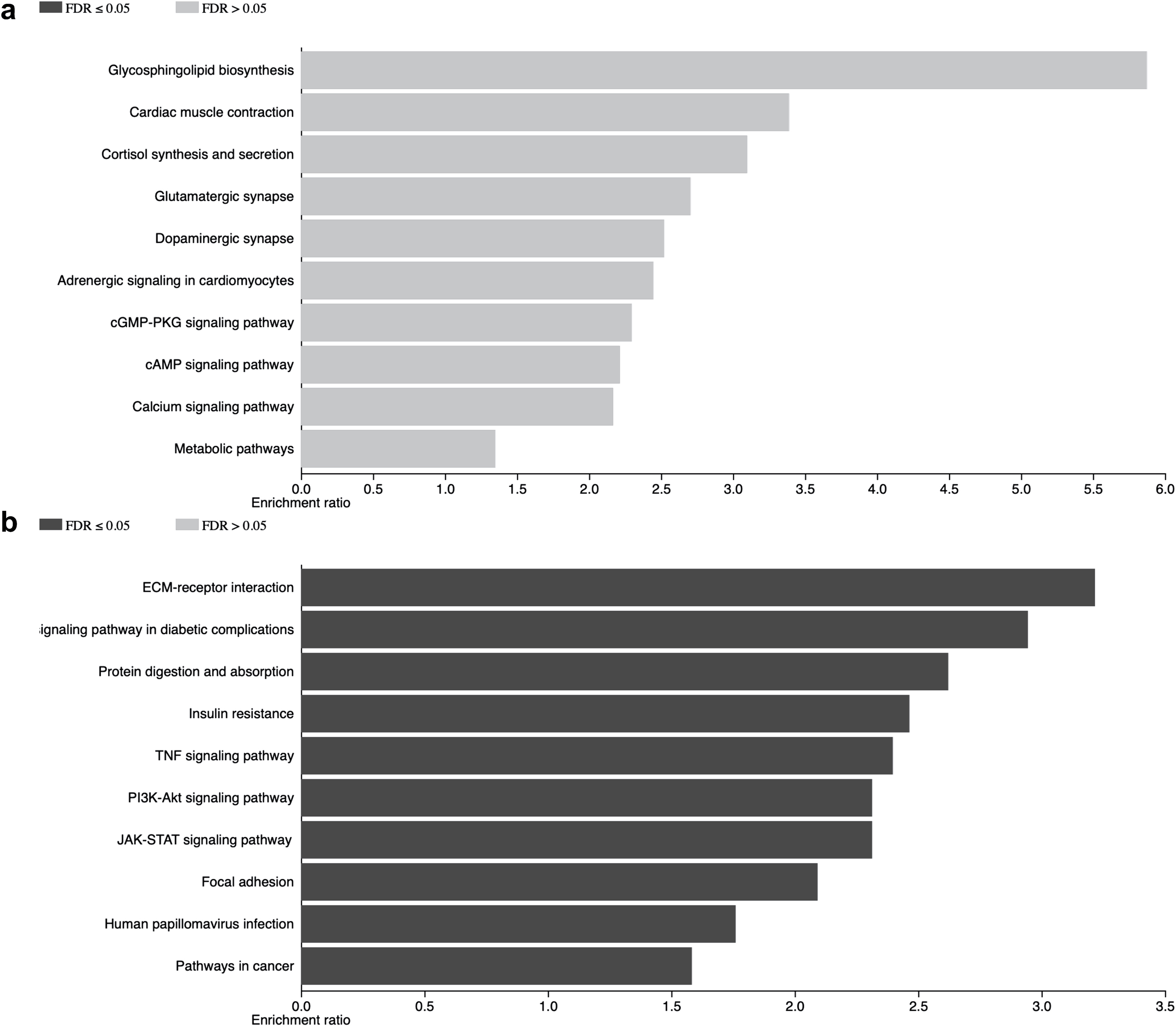
Displays the network interaction results. Panel a. Depicts the networks interactions, the largest node is comprised by ion metabolism-related events where Ca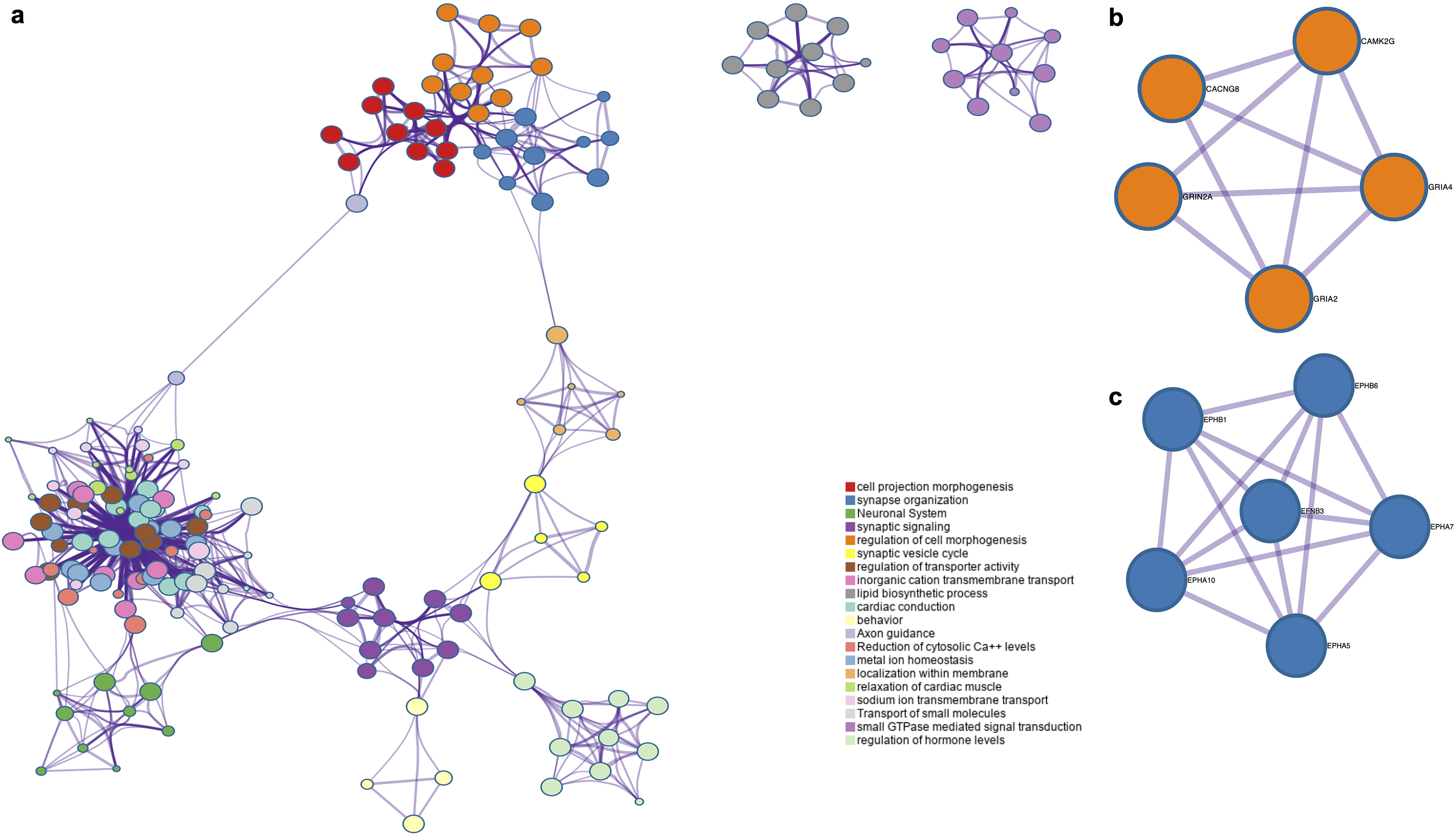
The upregulated genes found in GNFPA included coding genes such as CACNA2D4, SLC10A4, KLF8, PITX2, GATA3, SLC32A1, IDH1, CX3CR1, IL17D, KCNQ5, LEP-R, and SCN2B, as well as non-coding genes such as LINC01616, LINC00672, LINC00511, and MiR181A2HG (Fig. 3). The down-regulated genes included coding genes such as GH1, CSH1-2, POMC, GHRH-R, TSHB, and IL-6 as well as non-coding genes such as LINC00632, LINC01105, and MiR503HG.
The genes found to be upregulated are involved in different cellular pathways, including calcium signaling (CAMK2G and ATP2B2), cardiac muscle-related events (CACNG8 and SLC8A1), metabolic pathways (IDH1 and CYP11A1) as well as cell adhesion (CDH2, SDC2, and CADM1). The cellular pathways in which the down-regulated genes participate included insulin signaling (IRS2 and PCK1), signal transduction of growth factors (PRL-R, JAK1, STAT3) as well as TNF-related immune response and inflammation (CCL2 and CXCR4) (Fig. 4). Validations of these results are needed.
Upon identification of the up-regulated genes in GNFPA we carried out networks analysis to identify potential interactions between the different cellular processes altered in these neoplasias. The most notorious network was the one associated with ion-related events, such as the reduction of intracellular Ca
Protein-protein interactions amongst up-regulated genes
The up-regulated genes list was used to identify potential protein-protein interactions (PPI) in order to understand intracellular signaling and communication. Consistent with the previous results, where ion-related events are altered, we observed calcium related proteins (CAM2KG and CACNG8) interacting with glutamate receptors (GRIA2, GRIA4 and GRIN2A) (Fig. 5). Ephrin (EFNB3) and Ephrin receptors (EPHA10, -A7, -A5, -B6 and -B1) PPI was observed among others (Fig. 5).
Discussion
The present in silico analysis focuses on the molecular alterations in NFPA, which are the second most common pituitary lesion and the identification of new potential molecular markers. Currently we are collecting NFPA tissue, creating our own cohort, increasing the number of tissue samples to be analyzed to corroborate the present findings.
Interestingly, null cell adenomas and gonadotrope cell adenomas transcriptomes resemble remarkably and sharing most of the up- and down-regulated genes [7]. This allows us to suggest two potential hypotheses regarding the origin of these neoplasms. The first one is that NCNFPA originate in the non-differentiated stem cells from the pituitary gland, hence the negative expression of hormones commonly found in the cells of the pituitary gland. And the second is that the NCNFPA originate from gonadotropes NFPA and along the way they lose the transcriptional regulation for the expression of LH and FSH genes. Genes such as PITX2 and KLF8 that have been related to pituitary stem cell and cancer stem cells, respectively [8, 9], were found to be expressed in the gene sets obtained in our analysis. There is also evidence supporting the hypothesis that the null cell adenomas could share cell origin with the gonadotrope adenomas or that they could be the same entity [10]. Evidence supporting both of our hypotheses exists; therefore, there is the need to carry out more molecular characterization of a large number of PA. A third potentially viable scenario would be that null cell adenomas simply represent hormone-negative gonadotrope-cell adenomas, since their biological and clinical behavior is basically the same, in terms of invasiveness and recurrence rate. Although null-cell adenomas were once thought to behave more aggressively than gonadotrope-cell adenomas this has not been validated in more recent, larger-scale studies [11, 12]. Thus, our transcriptomic results are in concordance with clinical observations.
The transcriptome of PA showed clear alterations in calcium metabolism related genes. Calcium ion plays a central role in biological systems since it is an active participant in most intracellular signaling communication processes, from hormone signal transduction to immune-related events, apoptosis and control of cellular proliferation, and even epigenetic events [13]. Calcium metabolism also has been shown related to differentiation, migration, and invasion [14]. It also could represent a potential targets for molecular-based therapies. Currently, there are several drugs targeting Ca
Our results also point to an eminent role of immune-response related genes that could participate in PA biology. In this regard, there is evidence that several cytokines and chemokines could participate in the formation, establishment and/or progression of PA. They could act directly or indirectly in the pituitary cell function by altering the cell proliferation and hormone secretion among other processes [17, 18]. There is also evidence that Ca
In conclusion, there is a large requirement and an urgent need to molecularly characterize the cellular subtypes that comprise PA. The theories we propose based on our in silico analysis, particularly those alluding to the origin of null cell adenomas await further experimental proof using emergent molecular techniques such as single cell sequencing.
Footnotes
Acknowledgments
DMR is a recipient of the National Council for Science and Technology “Catedra CONACyT” program. This work was partially supported by grants 289499 from Fondos Sectoriales Consejo Nacional de Ciencia y Tecnologia Mexico and R-2015-785-015 from Instituto Mexicano del Seguro Social (MM).
Conflict of interest
The authors declare that they have no conflict of interest.