
Abstract
BACKGROUND:
Lung adenocarcinoma (LUAD) accounts for a significant proportion of lung cancer and there have been few diagnostic and therapeutic targets for LUAD due to the lack of specific biomarker. The aim of this study was to identify key long non-coding RNAs (lncRNAs) for LUAD.
METHODS:
The lncRNA and mRNA expression profiles of a large group of patients with LUAD were obtained from The Cancer Genome Atlas (TCGA). The differentially expressed lncRNAs (DElncRNAs) and mRNAs (DEmRNAs) were identified. The optimal diagnostic lncRNA biomarkers for LUAD were identified by using feature selection procedure and classification model. We established classification models including random forests, decision tree and support vector machine to distinguish LUAD and normal tissues. The lncRNAs-mRNAs co-expression networks and module identification were established by weighted gene co-expression network analysis (WGCNA). Functional annotation of pink and green modules was performed. The expression of selected DElncRNAs were validated by qRT-PCR.
RESULTS:
A total of 1364 DEmRNAs (468 down-regulated and 896 up-regulated mRNAs) and 260 DElncRNAs (88 down-regulated and 172 up-regulated lncRNAs) between LUAD and normal tissue were obtained. LANCL1-AS1, MIR3945HG, LINC01270, RP5-1061H20.4, BLACAT1, LINC01703, CTD-2227E11.1 and RP1-244F24.1 were identified as optimal diagnostic lncRNA biomarkers for LUAD. The area under curve (AUC) of the random forests model, decision tree model and SVM model were 0.999, 0.937 and 0.999, and the specificity and sensitivity of the three model were 98.3% and 99.8%, 93.2% and 99% and 100% and 98.4%, respectively. Co-expression networks analysis showed that RP11-389C8.2, CTD-2510F5.4 and TMPO-AS1 were co-expressed with 44, 242 and 241 mRNAs, respectively. Cell cycle, DNA replication and p53 signaling pathway were three significantly enriched pathways. The qRT-PCR results were consistent with our integrated analysis, generally. The GSE32863 and GSE104854 validation was consistent with our integrated analysis, generally.
CONCLUSION:
Our study identified eight DElncRNAs as potential diagnostic biomarkers of LUAD. Functional annotation of green module provided new evidences for exploring the precise roles of lncRNA in LUAD.
Keywords
Introduction
Lung cancer is the one of the leading causes of cancer-related mortality worldwide [1]. Among all lung cancers, more than 85% are categorized as non-small cell lung cancer, of which lung adenocarcinoma (LUAD) is one of the most common types of non-small cell lung cancer [2]. Since LUAD is easy to form metastases at an early stage, the prognosis for patients with LUAD is poor, with a particularly low 5-year survival rate for patients suffering from this disease at its advanced stages [3]. In spite of advancements that have been made for the diagnosis and treatment of LUAD, the 5-year overall survival of patients with LUAD remains low. Hence, it is urgently required to identify accurate indicators in the diagnostic and therapeutic targets of LUAD.
Emerging evidence has disclosed that deregulation of lncRNAs has been well recognized in cancer, and has been suggested to modulate cancer development at transcriptional and posttranscriptional levels [4, 5]. Some well-characterized lncRNAs have been reported to possess oncogenic or tumor suppressive roles and function as a biomarker for cancer diagnosis or prognosis [6, 7]. The emerging roles of lncRNAs in LUAD have also been investigated in previous studies. For instance, Su et al. demonstrated that LINC00472 contributed to the increase in LUAD cell apoptosis and the inhibition of proliferation [8]. Tian et al. showed that FENDRR and LINC00312 had diagnostic value in patients with LUAD [9]. LncRNA lOC100132354 promotes angiogenesis through VEGFA/VEGFR2 signaling pathway according to Wang et al.’s study on LUAD [10]. However, research for lncRNA underlying biomarkers in LUAD is rarely. Thus, identification of LUAD-related lncRNAs, and investigation of their molecular mechanisms are essential for understanding the occurrence and development of LUAD.
In the current study, we obtained the lncRNA and mRNA expression data of a large number of patients with LUAD from The Cancer Genome Atlas (TCGA), and attempted to identify the optimal diagnostic lncRNA biomarkers by using feature selection procedure and classification model. The mRNAs-lncRNAs co-expression networks and module identification were established by WGCNA. The functions of the potential lncRNA diagnostic biomarkers in LUAD were further analyzed by functional annotation of pink and green modules. To our knowledge, this is first time to find key lncRNAs in LUAD by using machine learning and weighted gene co-expression network analysis (WGCNA).
Materials and methods
Integrated profiles in TCGA
The Cancer Genome Atlas (TCGA) (
Identification DElncRNAs and DEmRNAs between LUAD and normal tissues
The undetectable lncRNAs and mRNAs (with read count value
Identification of optimal diagnostic lncRNA biomarkers for LUAD
To identify optimal diagnostic lncRNA biomarkers for LUAD, we performed feature selection procedures as follows. LASSO algorithm was conducted by the glmnet package to decrease dimensions of the data. We performed single10-fold cross-validation cycles with the coordinate descent algorithm for each fold and found regularization parameters that result in the smallest average mean squared errors across all folds. The optimal DElncRNAs were selected in LUAD and normal tissue.
To further identify the optimal diagnostic lncRNA biomarkers for LUAD, we performed feature selection procedures as follows. (1) The importance value of each lncRNA was ranked according to mean decrease in accuracy from large to small by random forest algorithm. (2) The optimum number of features was found by adding a DElncRNA at a time in the top down forward-wrapper packaging method. (3) By using support vector machine (SVM) at each increment and the optimal diagnostic lncRNA biomarkers were identified for LUAD.
The ‘random Forests’ packet (
Construction of the WGCNA coexpression network
WGCNA is an algorithm used to build co-expression modules and network. Briefly, a co-expression similarity matrix was calculated by the Pearson’s correlation coefficient between mRNAs and lncRNAs pairs. According to the scale-free topology criterion described in the WGCNA package documents, the similarity matrix was raised to a soft thresholding power of power
Functional annotation of green and pink module
To display the biological functions and the potential pathways of green and pink module for LUAD, Gene Ontology (GO) classification and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were performed by using g: Profilter (
Confirmation by qRT-PCR
According to the results of TCGA integration analysis, eight optimal diagnostic lncRNAs biomarkers (LANCL1-AS1, MIR3945HG, LINC01270, RP5-106- 1H20.4, BLACAT1, LINC01703, CTD-2227E11.1 and RP1-244F24.1) were screened as candidate lncRNAs. Ten tissues samples of LUAD patients (
Primer sequences used for qRT-PCR
Primer sequences used for qRT-PCR
The heat-maps of DElncRNAs and top 100 DEmRNAs between LUAD and normal tissues. (A) DEmRNAs. (B) DElncRNAs. Row and column represented DElncRNAs/DEmRNAs and tissue samples, respectively. The color scale represented the expression levels.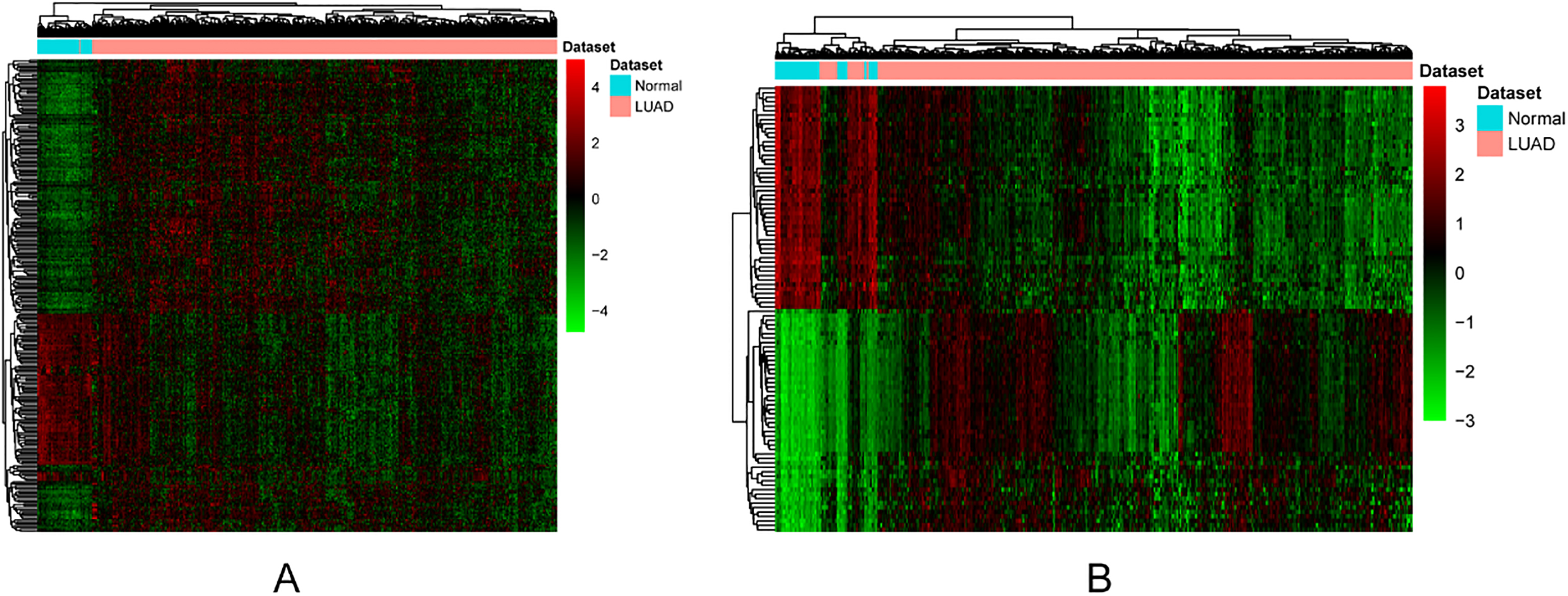
Identification of optimal lncRNA biomarkers for LUAD. (A) The importance value of each DElncRNA ranked according to the mean decrease in accuracy by using the random forest analysis. (B) The variance rate of classification performance when increasing numbers of the predictive DElncRNAs. (C) Heat-map of LUAD-specific eight lncRNAs biomarkers (LANCL1-AS1, MIR3945HG, LINC01270, RP5-1061H20.4, BLACAT1, LINC01703, CTD-2227E11.1 and RP1-244F24.1). (D–K) Box-plot displayed the expression levels of eight lncRNAs biomarkers between LUAD and normal tissues. The X-axis represented normal and LUAD groups. The Y-axis represented gene expression levels.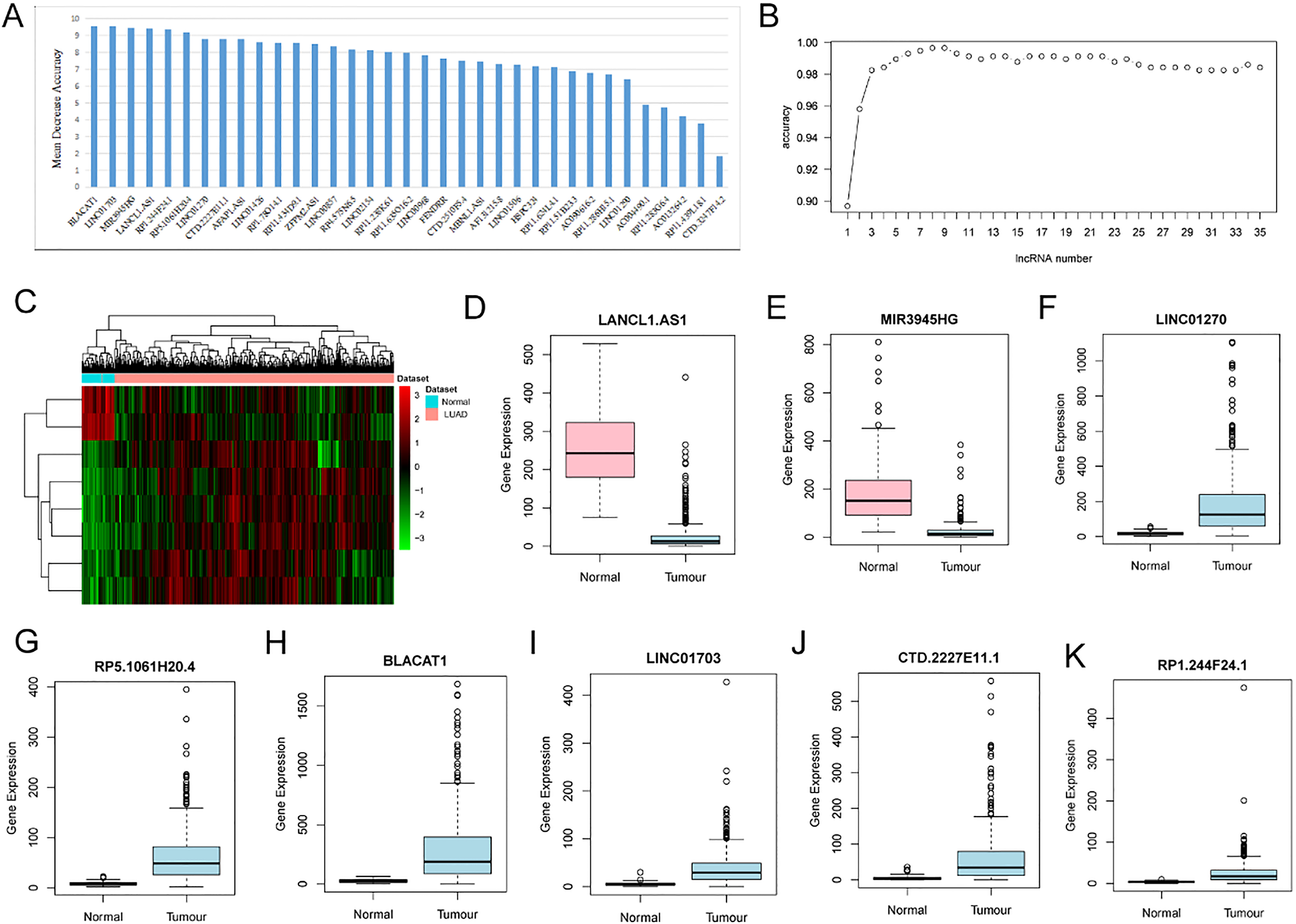
GSE32863 dataset was downloaded from the GEO (
Results
DEmRNAs and DElncRNAs in LUAD
The clinical data of 522 LUAD patients were downloaded from TCGA, among which, 513 LUAD patients were enrolled in this study. Males and females accounted for 46.2% and 53.8%, respectively. The medial age of these 513 patients was 65 years old. Whites, Asians and Black or African American accounted for 86.7, 1.6% and 11.7%, respectively. The tumor stages were as follows: Stage I, 54.2%; Stage II, 24%; Stage III, 16.7%; and Stage IV, 5.1%. The mRNA and lncRNA expression profiles of 513 LUAD tumor tissues and 59 normal tissues were obtained. A total of 3177 lncRNAs and 15183 mRNAs were available for analysis after removing those hardly detected lncRNAs and mRNAs.
A total of 1364 DEmRNAs (468 down-regulated and 896 up-regulated mRNAs) and 260 DElncRNAs (88 down-regulated and 172 up-regulated lncRNAs) between LUAD and normal tissue were identified with FDR
DElncRNAs between LUAD and normal tissues after reduced dimensions of data
DElncRNAs between LUAD and normal tissues after reduced dimensions of data
ROC analysis of eight LUAD-specific lncRNAs biomarkers. The ROC results of these eight diagnostic lncRNAs biomarkers (LANCL1-AS1, MIR3945HG, LINC01270, RP5-1061H20.4, BLACAT1, LINC01703, CTD-2227E11.1 and RP1-244F24.1) their combination based on support random forest (Fig. 3A), decision tree model (Fig. 3B) and SVM model model (Fig. 3C) and individual LANCL1-AS1 (Fig. 3D), MIR3945HG (Fig. 3E), LINC01270 (Fig. 3F), RP5-1061H20.4 (Fig. 3G), BLACAT1 (Fig. 3H), LINC01703 (Fig. 3I), CTD-2227E11.1 (Fig. 3J) and RP1-244F24.1 (Fig. 3K).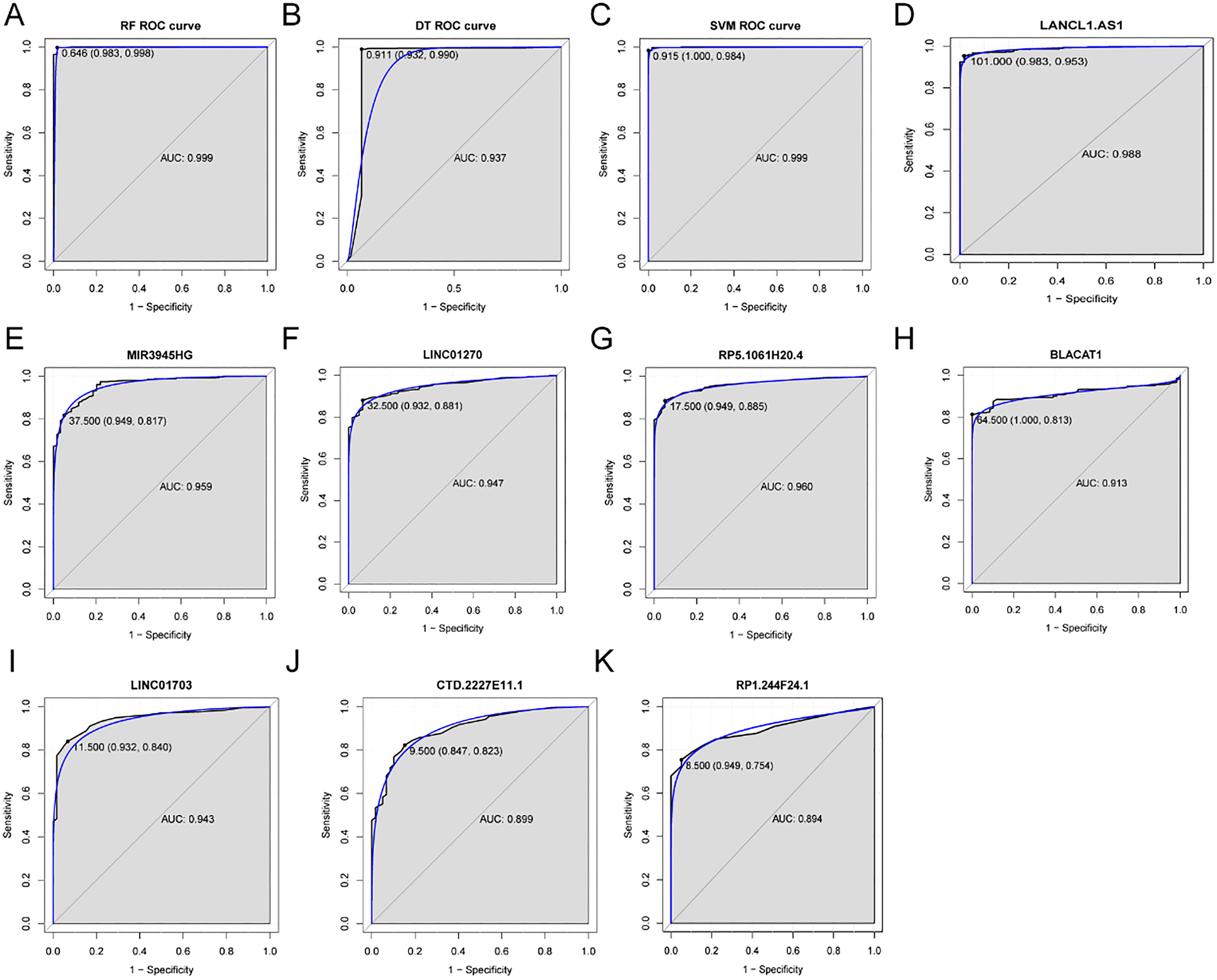
Network analysis of gene expression in LUAD identifies distinct modules of co-expression data. (A) Cluster dendrogram of the identifed lncRNA-mRNA co-expression modules. The lower panel shows colors designated for each module. Note that the leaves presented in the color gray indicates unassigned lncRNAs and mRNAs. (B) The cluster dendrogram of module eigengenes.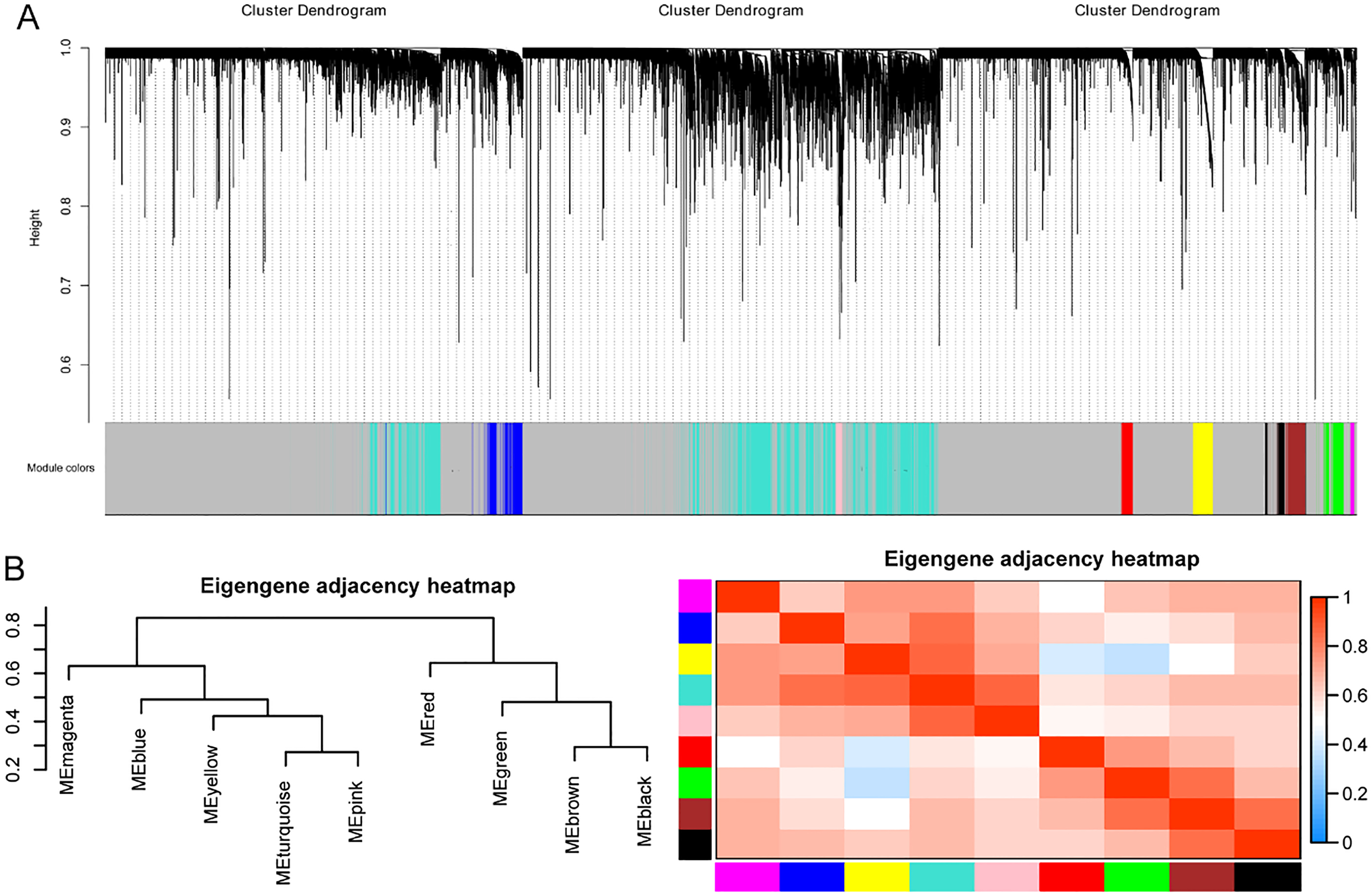
lncRNAs-mRNAs co-expression network. The ellipses and rhombuses were represented the mRNAs and lncRNAs, respectively. Green and pink color represented green module and pink module, respectively. The black border indicates DEmRNAs and DElncRNAs.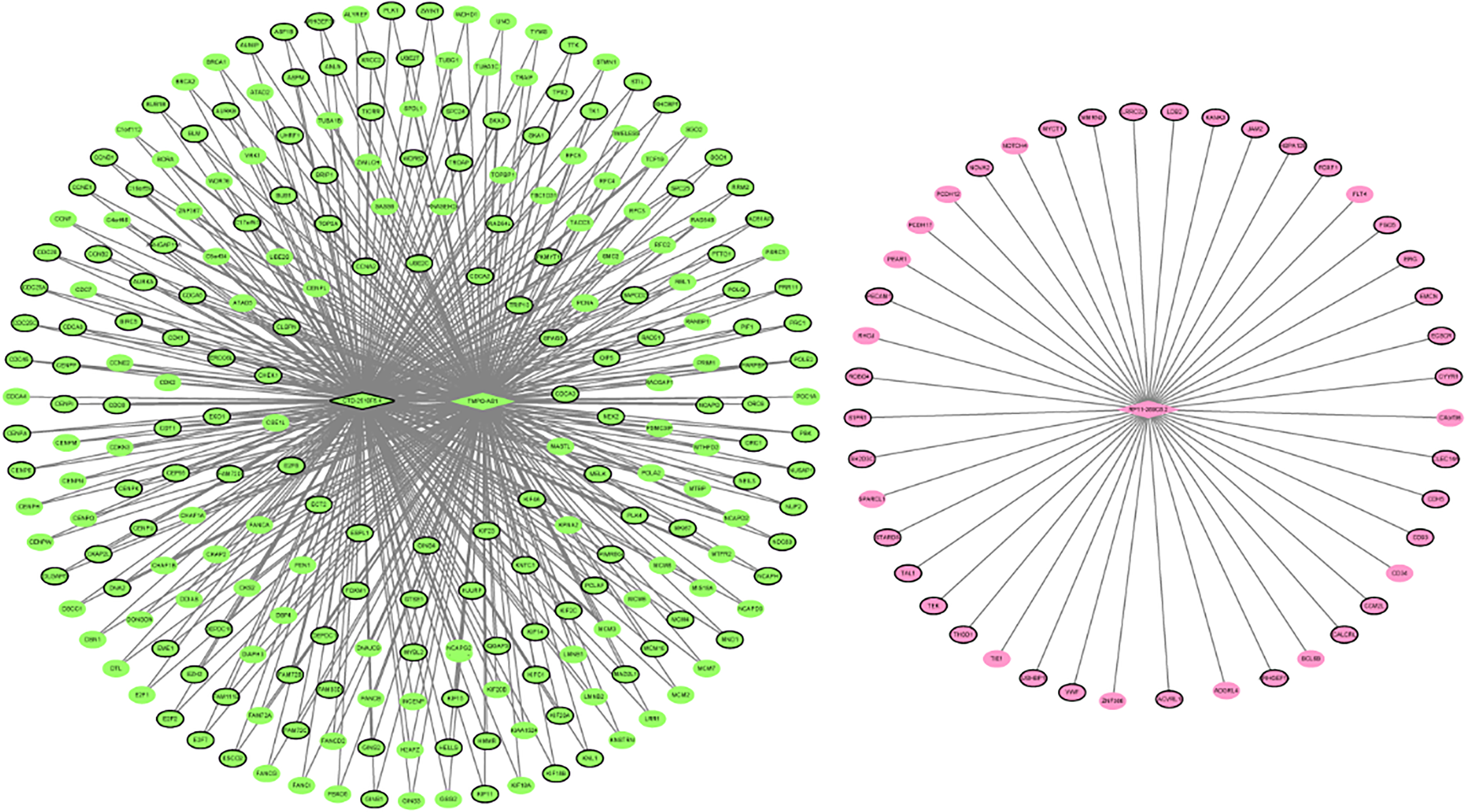
Based on reduced dimensions of the data, we obtained 35 DElncRNAs between LUAD and adjacent normal tissues by using LASSO algorithm (Table 2). The random forest analysis was used to rank the 35 DElncRNAs according to the mean decrease in accuracy (Fig. 2A). Ten-fold cross-validation result suggested that the average accuracy rate of 8 DElncRNAs (LANCL1-AS1, MIR3945HG, LINC01270, RP5-1061H20.4, BLACAT1, LINC01703, CTD-2227- E11.1 and RP1-244F24.1) reached the higher score for the first time (Fig. 2B). Therefore, these 8 DElncRNAs were determined as the optimal diagnostic lncRNA biomarkers for LUAD which were used to establish the random forests, decision tree and SVM models. The heat-map of these 8 DElncRNAs between LUAD and normal tissue were displayed in Fig. 2C. Box-plot uncovered the expression levels of these 8 DElncRNAs between LUAD and normal tissues (Fig. 2D–K). The AUC of the random forests model was 0.999 and the specificity and sensitivity of this model were 98.3% and 99.8%, respectively (Fig. 3A). The AUC of the decision tree model was 0.937 and the specificity and sensitivity of this model were 93.2.0% and 99%, respectively (Fig. 3B). The AUC of the SVM model was 0.999 and the specificity and sensitivity of this model were 100% and 98.4% (Fig. 3C). The AUC of all these 8 lncRNAs LANCL1-AS1, MIR3945HG, LINC01270, RP5-1061H20.4, BLACAT1, LINC01703, CTD-2227E11.1 and RP1-244F24.1 were also above 0.89 (Fig. 3D–K). Taken together, the AUC of all these eight lncRNAs and their combination were all greater than 0.89 which indicated the LANCL1-AS1, MIR3945HG, LINC012- 70, RP5-1061H20.4, BLACAT1, LINC01703, CTD-2227E11.1 and RP1-244F24.1 and their combination were related to LUAD and could predict the occurrence of LUAD.
lncRNAs-mRNAs co-expression networks and module identification
WGCNA was performed to identify lncRNAs- mRNAs co-expression networks and module identification. We identified nine module eigengenes (ME), including MEmagenta, MEblue, MEyellow, MEtur- quoise, MEpink, MEred, MEgreen, MEbrown and MEblack (Fig. 4A). In order to further explore the correlation between modules and tumor-adjacent cancer, the correlation analysis was performed, and the modules with the correlation coefficient
The function annotation of pink module. The x-axis shows-log FDR and y-axis shows GO terms or KEGG pathways.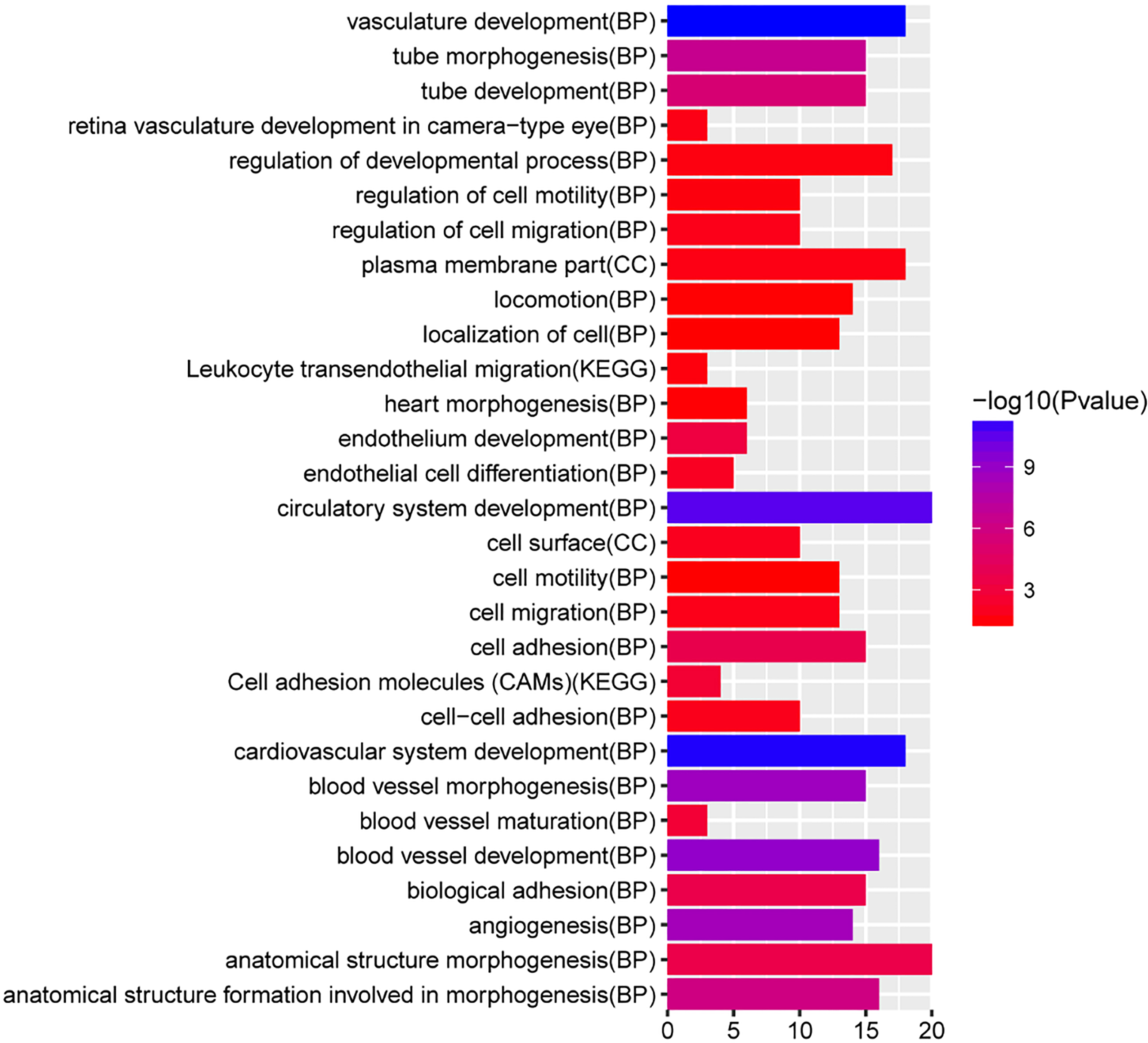
The function annotation of green module. The x-axis shows-log FDR and y-axis shows GO terms or KEGG pathways. (A) Biological process. (B) Cellular component. (C) Molecular function. (D) KEGG pathways.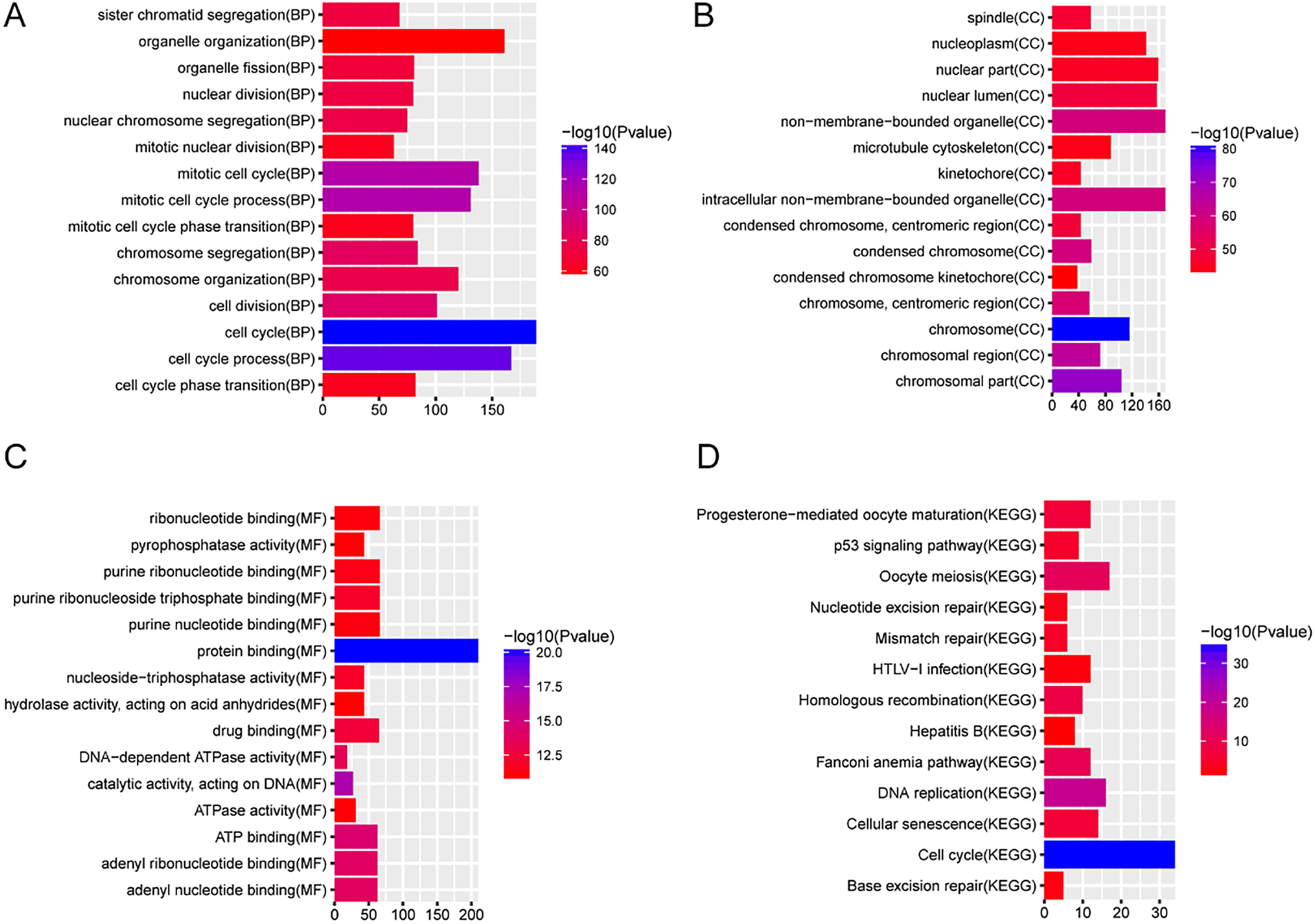
According to the functional annotation of 44 mRNAs in pink modules (Fig. 6), blood vessel morphogenesis (2.08E-09), angiogenesis (
Confirmation by qRT-PCR
We performed the confirmation of eight optimal diagnostic lncRNAs biomarkers by qRT-PCR. Based on TCGA, LANCL1-AS1 and MIR3945HG were down-regulated while the other six lncRNAs (LINC01270, RP5-1061H20.4, BLACAT1, LINC01703, CTD-2227- E11.1 and RP1-244F24.1) were up-regulated in LUAD compared to adjacent tissues. According to the qRT-PCR results, except for RP5-1061H20.4 and LINC- 01703, two lncRNAs were down-regulated and four lncRNAs were up-regulated which was consistent with the results of TCGA, generally (Fig. 8).
Validation optimal lncRNA biomarkers in LUAD tissue by qRT-PCR. 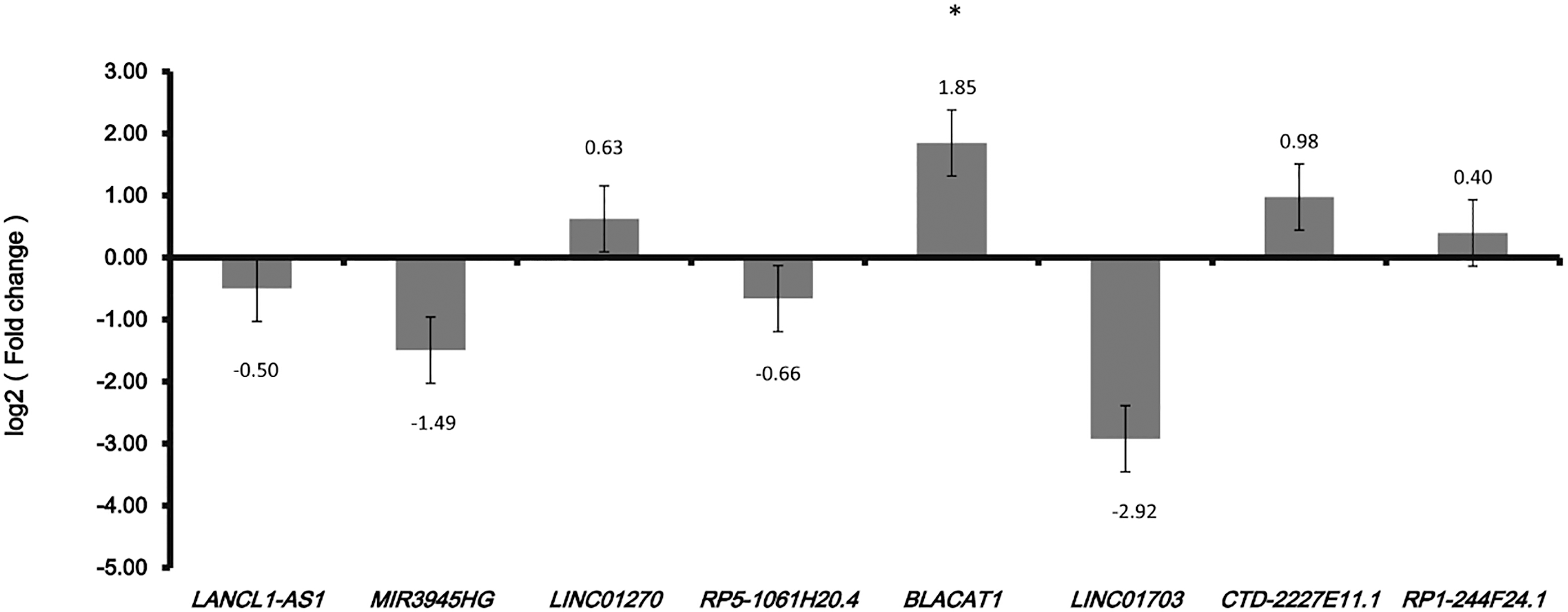
Validation of selected DEmRNAs and DElncRNAs in GSE32863 and GSE104854. The x-axis shows healthy normal control and LUAD groups and y-axis shows a log2 transformation to the intensities.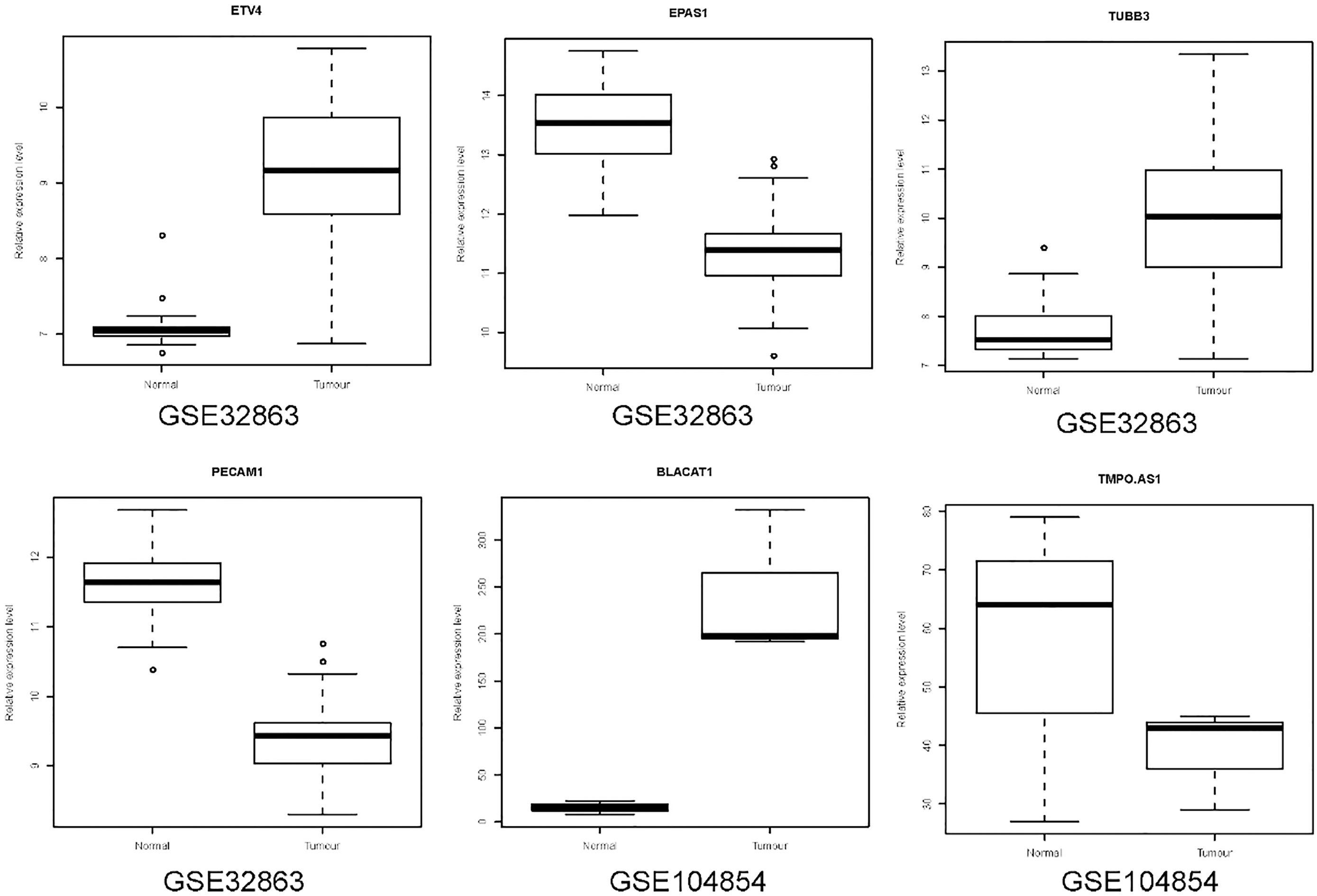
The expression pattern of selected DEmRNAs (ETV4, EPAS1, TUBB3 and PECAM1) and DElncRNAs (BLACAT1 and TMPO.AS1) was verified using GSE32863 and GSE104854 dataset, respectively. As shown in Fig. 9, TMPO.AS1 was down-regulated, which was inconsistent with our integration results. ETV4, TUBB3 and BLACAT1 were up-regulated while EPAS1 and PECAM1 were down-regulated in LUAD, which was consistent with our integration results, suggesting that the results were convincing.
Discussion
Lung cancer has the highest morbidity and mortality rate of lethal disease worldwide, and LUAD is one of the most common types of lung cancer [13]. Although the prognosis and diagnosis of LUAD has been achieved through studies, the 5-year overall survival of patients with LUAD remains low, requiring continued research to identify novel biomarkers and decipher the detailed molecular mechanisms underlying of LUAD. LncRNAs have been verified to play a key role in the occurrence and progression of lung cancer [1, 8]. Here is a report studying the DEmRNAs and DElncRNAs in a large group of patients with LUAD from TCGA. A total of 1364 DEmRNAs (468 down-regulated and 896 up-regulated mRNAs) and 260 DElncRNAs (88 down-regulated and 172 up-regulated lncRNAs) between LUAD and normal tissue were obtained. Additionally, eight optimal diagnostic lncRNAs biomarkers for LUAD were identified by feature selection procedure and classification model assay. Moreover, we used the WGCNA to build lncRNAs-mRNAs co-expression networks and module identification. The functional annotation of pink and green modules were evaluated.
Feature selection methods provided by machine learning approaches are an interesting, flexible and robust alternative for identifying predictors that contribute to disease occurrence [14]. Compared with similar type of LUAD studies [15, 16, 17], we mainly used the machine learning to identify optimal diagnostic lncRNA biomarkers of LUAD. The AUC of all these eight lncRNAs (LANCL1-AS1, MIR3945HG, LINC01270, RP5-1061H20.4, BLACAT1, LINC01- 703, CTD-2227E11.1 and RP1-244F24.1) and their combination in LUAD were more than 0.89, which indicated eight lncRNAs and their combination could predict the occurrence of LUAD. To our knowledge, except MIR3945HG and BLACAT1, one down-regulated DElncRNA (LANCL1-AS1) and five up-regulated DElncRNA (LINC01270, RP5-1061H20.4, LINC01703, CTD-2227E11.1 and RP1-244F24.1) in LUAD was firstly reported and their biological function remain unclear.
It is reported that MIR3945HG was identified as novel candidate diagnostic markers for tubercu- losis [18]. Recent research results indicated that MIR3945HG was the highest diagnostic values biomarker in the diagnosis of lung squamous cell carcinoma, and was closely related to the survival time of lung squamous cell carcinoma [15]. In the present study, we found that MIR3945HG was down-regulated in both informatics and qRT-PCR validation. Therefore, we hypothesized that MIR3945HG might involve in the development of LUAD.
BLACAT1 (bladder cancer associated transcript 1), also known as linc-UBC1, is a novel identified lncRNA in bladder cancer, and has been tested to function as an oncogenic lncRNA in several types of human cancer [19, 20, 21]. The overexpression of BLACAT1 in non-small cell lung cancer tissue and cells, and discovered the oncogenic role of BLACAT1 in non-small cell lung cancer genesis through sponging miR-144, providing a potential biomarker for early detection and prognosis prediction of non-small cell lung cancer [22]. It is reported BLACAT1 was associated with the malignant status and prognosis in patients with small-cell lung cancer, and functions as an oncogenic lncRNA in regulating cell proliferation and motility, suggesting BLACAT1 might act as a potential target for small-cell lung cancer revention and treatment [23]. In this study, we found that BLACAT1 was the top DElncRNA in LUAD, which was consistent with reports of other researchers [24], indicating the TCGA integration analysis data were reliable.
WGCNA results showed that green was positively with associated with tumor-adjacent cancer and contained a total of 241 up-regulated mRNAs and 2 up-regulated lncRNAs (CTD-2510F5.4 and TMPO-AS1). In this study, CTD-2510F5.4 were significantly up-regulated in LUAD. Moreover, CTD-2510F5.4 was also found to be differentially expressed in another study that used RNA-seq data from TCGA and two independent experiments [1, 25], which supports the reliability of our results. Li et al. showed that TMPO-AS1 was revealed to be prognostic biomarker for LUAD [17]. Peng et al. reported that TMPO-AS1 could affect the prognosis of LUAD patients through regulating cell cycle and cell adhesion [16]. Herein, we found that TMPO-AS1 was significantly up-regulated in LUAD. Hence, we further confirmed that TMPO-AS1 might be prognostic biomarker for LUAD. According to the functional annotation of 241 mRNAs in green modules, cell cycle, DNA replication and p53 signaling pathway were three significantly enriched pathways. Therefore, we speculated that CTD-2510F5.4 and TMPO-AS1 might involve in the development of LUAD by regulating signaling pathway of cell cycle, DNA replication and p53 signaling pathway.
In summary, we identified 1364 DEmRNAs and 260 DElncRNAs in LUAD compared to normal tissues. The feature selection procedure and classification model assay was to obtain eight optimal diagnostic lncRNAs biomarkers for LUAD. WGCNA was used to identify mRNAs-lncRNAs co-expression networks, and we obtained three lncRNAs (RP11-389C8.2, CTD-2510F5.4 and TMPO-AS1), which were related to LUAD, and built lncRNAs-mRNAs co-expression networks. However, there are limitations to our study. Firstly, the sample size in the confirmation by qRT-PCR was small and large numbers of samples of LUAD are needed for further research. Secondly, these DEmRNAs and DElncRNAs in LUAD were identified and the bio-function was not studied. Therefore, in vivo and in vitro experiments were necessary to illuminate the biological roles of DEmRNAs and DElncRNAs in LUAD in the future work.