
Abstract
Objective
This study aimed to identify significantly differentially expressed genes (DEGs) related to cervical cancer by exploring extensive gene expression datasets to unveil new therapeutic targets.
Methods
Gene expression profiles were extracted from the Gene Expression Omnibus, The Cancer Genome Atlas, and the Genotype-Tissue Expression platforms. A differential expression analysis identified DEGs in cervical cancer cases. Weighted gene co-expression network analysis (WGCNA) was implemented to locate genes closely linked to the clinical traits of diseases. Machine learning algorithms, including LASSO regression and the random forest algorithm, were applied to pinpoint key genes.
Results
The investigation successfully isolated DEGs pertinent to cervical cancer. Interleukin-24 was recognized as a pivotal gene via WGCNA and machine learning techniques. Experimental validations demonstrated that human interleukin (hIL)-24 inhibited proliferation, migration, and invasion, while promoting apoptosis, in SiHa and HeLa cervical cancer cells, affirming its role as a therapeutic target.
Conclusion
The multi-database analysis strategy employed herein emphasized hIL-24 as a principal gene in cervical cancer pathogenesis. The findings suggest hIL-24 as a promising candidate for targeted therapy, offering a potential avenue for innovative treatment modalities. This study enhances the understanding of molecular mechanisms of cervical cancer and aids in the pursuit of novel oncological therapies.
Keywords
Introduction
Cervical cancer continues to be a major health issue globally, with a disproportionately high burden in developing countries, where it leads to a substantial number of cases and deaths.1–3 Despite advancements in the prevention and treatment of cervical cancer in recent years, such as the introduction of the national cervical screening program by the National Health Service in 1988, which has led to a reduction of over one-third of cases in England, 4 the malignant biological behaviors of the disease, including rapid proliferation, invasion, and migration, remain major challenges for treatment.3,5,6 Currently, surgery, radiotherapy, and chemotherapy are the primary treatment modalities, but these approaches often come with significant side effects and risks of recurrence.1,7–9 A retrospective study of cervical cancer patients treated at the Royal Marsden Hospital in the United Kingdom from 2004 to 2014 revealed that 70% of recurrent or metastatic cervical cancer patients who received systemic therapy subsequently underwent second-line treatment, with an overall response rate of 13.2%, a median progression-free survival of 3.2 months, and a median overall survival of 9.3 months. 10 This finding underscores the critical need for the development of more effective and safer therapeutic options. 11
On the basis of these findings, gene therapy represents a highly promising field of medical research, attracting substantial attention for its potential to provide targeted treatment options.12–14 Human interleukin-24 (hIL-24) is notable for its wide range of biological actions, including its tumor-suppressing effects in various cancer forms.15–17 Although research has shown the ability of hIL-24 to inhibit tumor cell growth and promote apoptosis, its use in treating cervical cancer is not well explored.18–20
To address this gap, our study investigates the specific impact of hIL-24 on SiHa and HeLa cervical cancer cells. We created a pcDNA3.1 (+)-hIL-24 recombinant plasmid and introduced it into SiHa and HeLa cells through lipofection. 21 We assessed the gene’s effect on cell proliferation using PCR and MTT assays 22 and evaluated its influence on cell invasion and migration employing Transwell assays.23,24
We performed a thorough examination of the inhibitory effects of hIL-24 on the growth of SiHa and HeLa cells and assessed its regulatory role in cell invasion and migration. The outcomes of this study are anticipated to provide new theoretical and practical groundwork for incorporation of hIL-24 into cervical cancer treatment strategies. By examining the mechanism of action of hIL-24, we aspire to amplify the effectiveness and safety of treatments available to cervical cancer patients, contributing to the advancement of the fields of oncology and gene therapy.
Materials and methods
This study does not involve human or animal experimentation and therefore did not require ethics committee approval.
Gene Expression Omnibus (GEO), The Cancer Genome Atlas (TCGA), and Genotype-Tissue Expression (GTEx) transcriptome data
We obtained transcriptome data from 309 cervical cancer patients, including 306 cervical cancer tissue samples and three adjacent normal tissue samples, from TCGA database (https://portal.gdc.cancer.gov/). Transcriptome data were downloaded from the GTEx database for 10 normal cervical tissues. The GEO database was accessed at https://www.ncbi.nlm.nih.gov/gds to obtain the GSE63514 and GSE192804 datasets. The GSE63514 dataset included 28 cervical cancer tissues and 24 normal cervical tissues, while the GSE192804 dataset consisted of six cervical cancer tissues and six normal cervical tissues.25,26
Differential gene screening
Differential mRNA expression was filtered using the ‘limma’ package in R (https://www.r-project.org). A P-value less than 0.05 was defined as the screening criterion for TCGA. In addition, the criteria of an absolute log2 fold change greater than 2 and a P-value less than 0.05 were used for further filtering. We generated a volcano plot using the ggplot2 R package developed by Hadley Wickham (http://had.co.nz/ggplot2/). The Xiantao Academic Database (https://www.xiantaozi.com/) was used to create a Venn diagram.27,28
Weighted gene co-expression network analysis (WGCNA)
The expression clustering and phenotype association analyses were conducted on gene data from TCGA database using the ‘WGCNA’ package in the R language. The most relevant gene modules associated with cervical cancer were identified, and the genes within these modules were extracted for further analysis. 29
Functional enrichment analysis
We performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses on the differentially expressed genes (DEGs) in the cervical cancer samples. The SangerBox database (http://sangerbox.com/home.html) was utilized for this analysis. In addition, we generated visualizations.30,31
Cell culture
SiHa and HeLa cells derived from human cervical cancer were procured from Beyotime Biotechnology Co., Ltd. (Shanghai, China). The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum and 1% (100×) penicillin-streptomycin. The culture conditions included a temperature of 37°C and 5% carbon dioxide.23,32
Plasmid extraction and siRNA sequence
The pcDNA3.1 (+)-hIL-24 plasmid was synthesized by Shanghai Biotechnology Co., Ltd. (Shanghai, China) and subsequently sequenced by Shanghai Gene Engineering Co., Ltd. (Shanghai, China). The plasmid extraction and purification reagents were purchased from Beyotime Biotechnology Co., Ltd. DH5α Escherichia coli cells transformed with the pcDNA3.1(+)-hIL-24 plasmid were inoculated into LB medium supplemented with ampicillin. The cultures were then incubated overnight at 37°C with shaking at 250 rpm. Plasmids were extracted according to the instructions provided by the plasmid extraction kit.33,34 The siRNAs targeting hIL-24 were as follows: #1: GCAAAGCCTGGGACTTTA and #2: CCAACAACTTTGTTCAT. 35 The negative control is denoted as siNC.
Cell transfection
The Lipo8000™ transfection reagent was purchased from Shanghai Beyotime Biotechnology Co., Ltd. Approximately 500,000 cells per well were seeded into a six-well plate the day before transfection and cultured in an antibiotic-free medium for 18 to 24 hours. The cells were cultivated until the following day, achieving a cell density of approximately 70% to 80%. During transfection, 2 mL of fresh medium without antibiotics was used to replace the medium in each well. Then, 2.5 μg of DNA was combined with 4 μL of Lipo8000™ transfection reagent in 125 μL of DMEM, and 125 μL of the mixture was added to each well, followed by a 48-hour incubation period. 36
Cell growth inhibition assay
The densities of transfected and non-transfected SiHa and HeLa cells were adjusted to 2.5 × 104 cells/mL in DMEM. Subsequently, the cells were seeded in a 96-well plate with 200 μL of cell suspension per well. Following a 72-hour incubation period at 37°C in 5% carbon dioxide, 20 μL of MTT solution was added to each well, and the plate was incubated for another 4 hours at 37°C. The culture medium was removed, 150 μL of dimethyl sulfoxide was added, and the sample was thoroughly mixed for 10 minutes. The absorbance of each well was measured at a wavelength of 568 nm using an instrument equipped with an enzyme label.37,38
Cell migration experiment
This study aimed to investigate the impact of hIL-24 on the migratory capacity of SiHa and HeLa cells using a Transwell assay. The transfected and untransfected SiHa and HeLa cells were centrifuged at 1000 rpm for 5 minutes. The supernatant was removed, and the cells were washed twice with phosphate-buffered saline (PBS). The cells were then transferred to DMEM and resuspended. The cell concentration was adjusted to 3 × 105/mL using a hemocytometer. Then, 800 μL of DMEM with 10% penicillin-streptomycin was added to a 24-well plate, and a Transwell chamber was placed inside. A 200-μL cell suspension was inoculated into the upper chamber of the Transwell system for each group, and the plate was incubated at 37°C for 48 hours. Afterward, the insert was removed, and the chamber was washed with PBS. Cell fixation was performed by incubating the cells in a 70% ethanol solution for 1 hour, followed by staining with a 0.5% crystal violet solution at room temperature for 20 minutes. Following washing with PBS, a clean cotton swab was used to remove the non-migrated cells on one side of the chamber. The cells were observed and photographed under a microscope (BX63, Olympus, Japan).39–42
Cell invasion assay
Before the experiment, Matrigel was melted and diluted with serum-free medium in a ratio of 1:3, and The Transwell chamber was placed in a 24-well plate. Next, 100 μL of diluted matrix gel was applied evenly onto the polycarbonate membrane at the bottom of each well. The plate was then incubated at 37°C in a 5% carbon dioxide incubator for 30 minutes. The subsequent steps were identical to those of the cell migration experiment.43–45
Apoptosis assessment
The transfected SiHa and HeLa cells were centrifuged at 1200 rpm for 5 minutes. After centrifugation, the supernatant was removed, and the cells were resuspended in PBS. The cells were then washed twice using sterile PBS and centrifuged at 1200 rpm for 5 minutes. The cells were analyzed using flow cytometry, following the instructions provided by Shanghai BioCloud Biotechnology Co., Ltd. (Shanghai, China) for the Annexin V-FITC Apoptosis Detection Kit.46–48
Statistical analysis
The R language, version 4.2.1, was used for this study. Compilation of the R language was performed using the RStudio integrated development environment (current version 2022.12.0-353). Perl version 5.30.0 (https://www.perl.org/get.html) and GraphPad Prism version 8.0 software (https://www.graphpadchina.com/download.html) were used for file processing.
Statistical analysis was performed using SPSS 17.0 software (SPSS Inc., Chicago, IL, USA). After the normality of the data was assessed, multiple comparisons were conducted using analysis of variance. Inter-group comparisons were performed using t-tests. Data are represented as the mean ± standard deviation. A P-value less than 0.05 indicates statistical significance.49,50
Results
Identification of the essential genes for cervical cancer through a comprehensive analysis of the GEO, TCGA, and GTEx databases
To investigate the crucial genes involved in cervical cancer, we retrieved data from the GTEx database. We obtained transcriptome data for 31 diverse tissue types from both sexes (Figure 1). Through differential analysis, we identified 1804 genes that exhibited differential expression in cervical cancer compared with the adjacent normal cervical tissues. This analysis used transcriptomic data from TCGA and the GTEx databases (Figure 2(a)). Furthermore, by using the GSE63514 dataset, we obtained 510 genes that displayed differential expression in cervical cancer compared with normal cervical tissues (Figure 2(b)). Furthermore, we identified 1702 genes that exhibited differential expression in cervical cancer compared with normal cervical tissue using data from the GSE192804 dataset (Figure 2(c)). The results demonstrate our successful acquisition of transcriptome data from TCGA and the GTEx databases and the retrieval of two microarray datasets from the GEO database. Furthermore, differential analysis allowed us to individually identify distinct genes from each dataset.

Classification of transcriptional data from the Genotype-Tissue Expression (GTEx) database. (a) Classification of transcriptional data from the GTEx database by tissue type and (b) classification of transcriptional data from the GTEx database by sex and tissue type.
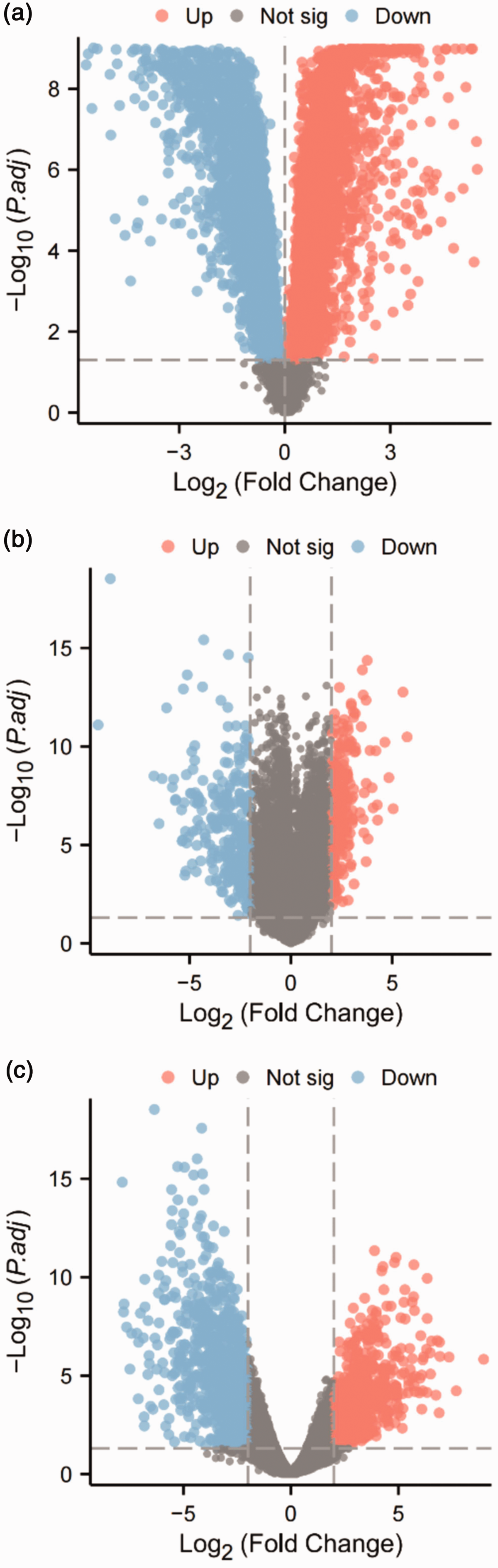
Identification of differentially expressed genes related to cervical cancer in the Gene Expression Omnibus (GEO), The Cancer Genome Atlas (TCGA), and the Genotype-Tissue Expression (GTEx) databases. (a) Volcano plot of differentially expressed genes between 13 normal cervical tissues and 306 cervical cancer tissues in TCGA and the GTEx databases. (b) Volcano plot of Continued.differentially expressed genes between 24 normal cervical tissues and 28 cervical cancer tissues in the GSE63514 dataset and (c) Volcano plot of differentially expressed genes between six normal cervical tissues and six cervical cancer tissues in the GSE192804 dataset. In the volcano plots, blue dots represent genes downregulated in cervical cancer, red dots represent genes upregulated in cervical cancer, and gray dots represent genes with no differences.
Use of WGCNA to uncover genes characteristic of cervical cancer: a thorough investigation of modules associated with the disease
To effectively identify disease-related gene characteristics closely associated with cervical cancer, we performed WGCNA using TCGA and the GTEx databases. We obtained nine gene modules: black, blue, brown, green, gray, magenta, red, turquoise, and yellow. Among these modules, the turquoise module exhibited the highest proportion of gene importance (Figure 3(a)). The correlation analysis results between the module genes and cervical cancer revealed a negative correlation between the turquoise module genes and cervical cancer. This result suggests that the turquoise module genes may exert inhibitory effects on cervical cancer (Figure 3(b)). To identify potential overlap, we conducted an intersection analysis between 988 disease-associated genes extracted from the turquoise module and the DEGs identified from the GEO, TCGA, and GTEx databases. This analysis led to the identification of six genes that intersected across these datasets (Figure 3(c)).
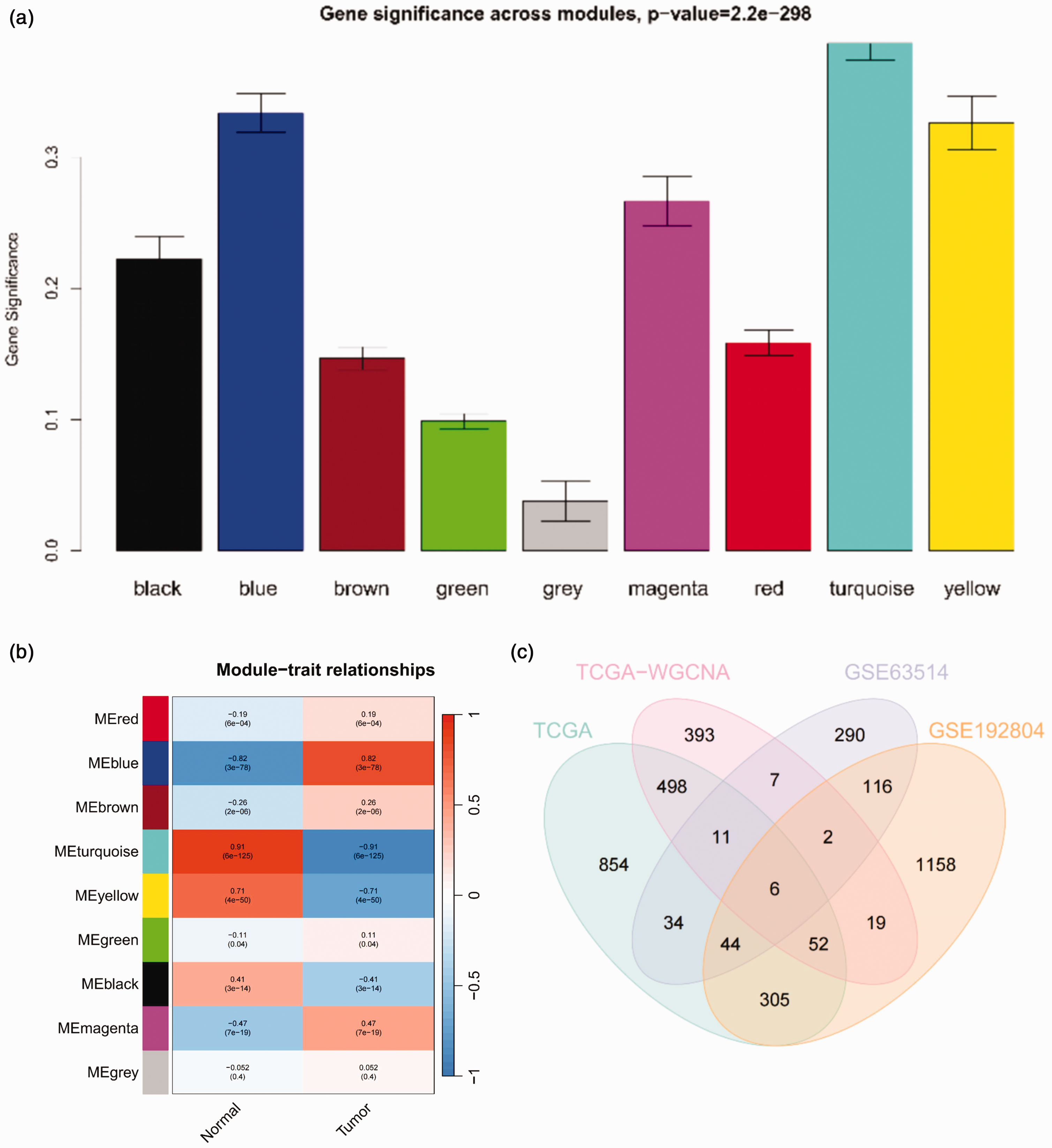
Weighted gene co-expression network analysis (WGCNA) results. (a) Graph showing the importance of module genes in cervical cancer according to WGCNA (N = 319). (b) WGCNA results of genes related to cervical cancer (N = 319) and (c) venn diagram showing the intersection of differentially expressed genes in cervical cancer samples from The Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression (GTEx) databases, genes from WGCNA analysis (turquoise module), and genes with differential expression in the GSE63514 and GSE192804 datasets from cervical cancer tissues.
Next, we performed GO and KEGG enrichment analyses on the six overlapping genes. GO enrichment analysis showed that the DEGs in the cervical cancer samples were primarily enriched in biological processes, including tissue development, extracellular structure organization, and wound healing (Figure 4(a)). The KEGG enrichment analysis revealed that the DEGs in the cervical cancer samples were primarily enriched in signaling pathways, including extracellular matrix-receptor interaction, human papillomavirus infection, and focal adhesion (Figure 4(b)). These results indicate that we identified six genes closely associated with cervical cancer through WGCNA and differential analysis.
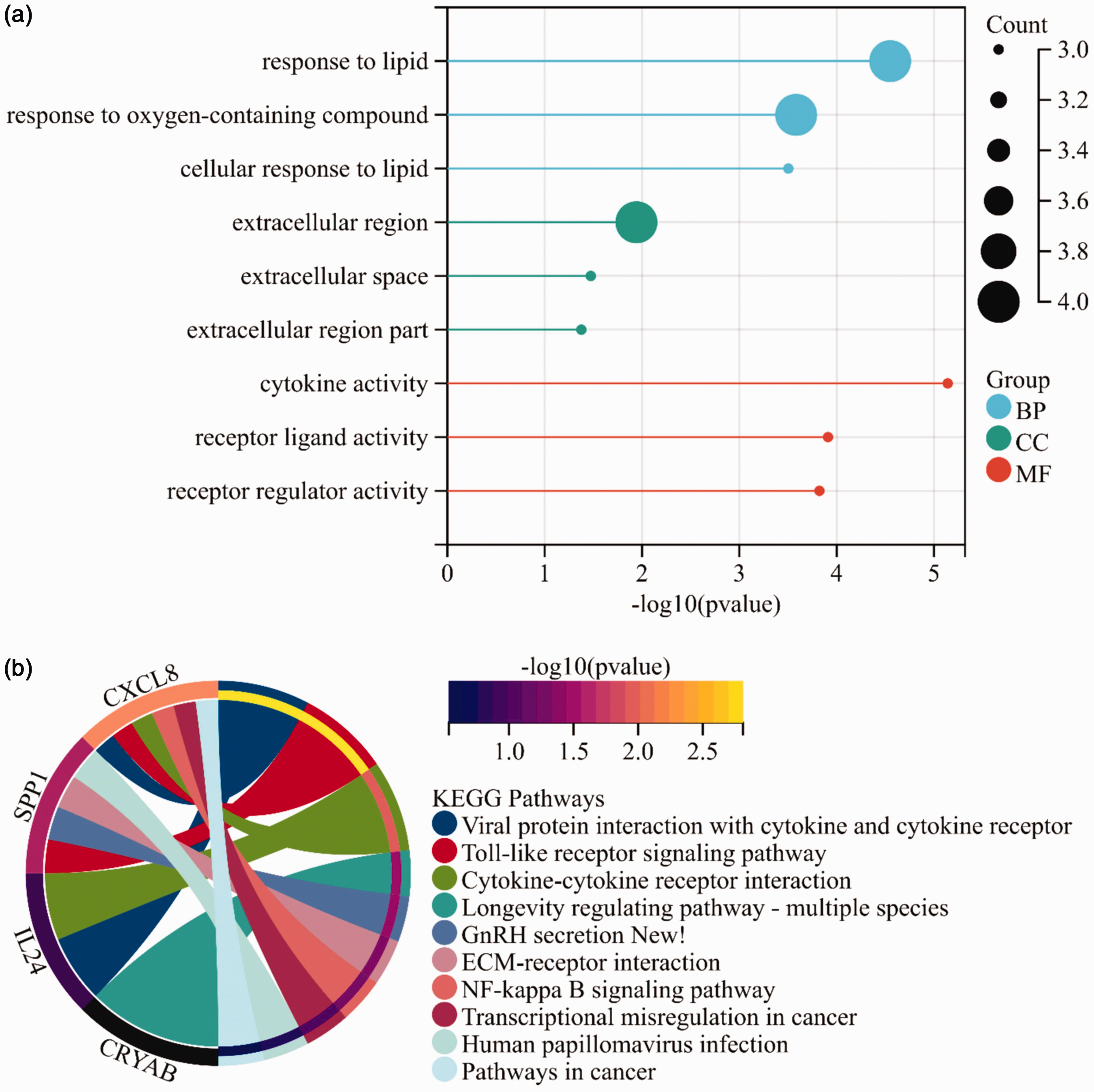
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis results. (a) Bar plot of GO enrichment for intersecting genes, with blue, green, and red representing the biological process (BP), cellular component (CC), and molecular function (MF) domains, respectively and (b) KEGG pathway circular diagram for intersecting genes.
Machine learning using the LASSO and random forest algorithms was vital for disease gene screening
Subsequently, the expression levels of the six genes in the GSE192804 dataset were extracted, and a multivariate Cox analysis with LASSO regression was conducted. This analysis identified five disease-associated genes, namely ACOX2, IL-24, SPP1, CRYAB, and ANKRD22 (Figure 5(a–b)). In addition, we used the random forest algorithm to assess gene importance, identifying IL-24 as a disease-associated gene (Figure 5(c)). Finally, by performing intersection analysis, we identified a single gene associated with cervical cancer, IL-24 (Figure 5(d)). Based on these results, we successfully identified the genes associated with cervical cancer.

A machine learning algorithm for screening disease-associated genes for cervical cancer. (a) LASSO analysis results, with the x-axis representing log(λ) values and the y-axis representing binomial deviance. The dashed line represents the log(λ) value corresponding to the optimal binomial deviance and the number of genes retained. (b) LASSO analysis results, with the x-axis representing the L1 Norm and the y-axis representing the regression coefficient. (c) Random forest algorithm result graph and (d) venn diagram showing the intersection of disease-associated genes selected by the LASSO regression and random forest algorithms, two machine learning algorithms.
hIL-24: a comprehensive study of the newly discovered multi-functional anti-cancer protein and its interaction with SiHa and HeLa cells
To further investigate the impact of hIL-24 on cervical cancer, we constructed a hIL-24 overexpression plasmid (pcDNA3.1 (+)-hIL-24) and designed siRNAs to knock down hIL-24, which were then introduced into SiHa and HeLa cervical cancer cells to evaluate their biological effects. MTT analysis revealed a significant decrease in the proliferative capacity of SiHa and HeLa cells in the pcDNA3.1 (+)-hIL-24 group compared with that in the transfection reagent and empty plasmid groups. Conversely, knockdown of hIL24 resulted in a significant enhancement of the proliferative capacity of SiHa and HeLa cells (P < 0.01), highlighting the crucial role of hIL-24 in inhibiting the growth of SiHa and HeLa cells (Figure 6(a), Figure 7(a)). Furthermore, the cell migration experiments indicated that hIL-24 significantly inhibited the migration abilities of SiHa and HeLa cells. Transfection of sihIL24 into SiHa and HeLa cells resulted in an enhanced migration ability compared with that in the negative control group transfected with siNC (P < 0.01) (Figure 6(b), Figure 7(b)). The invasion assay further confirmed that hIL-24 overexpression markedly suppressed the invasion capabilities of SiHa and HeLa cells. The pcDNA3.1(+)-hIL-24 plasmid group showed the lowest invasion ability, with an average of 84.3 Hela cells and 74.3 Siha cells. Compared with the negative control group transfected with siNC, the invasion abilities of SiHa and HeLa cells transfected with sihIL-24 were significantly increased (P < 0.01) (Figure 6(c), Figure 7(c)).

Statistical results of the effect of human interleukin-24 (hIL-24) on the biological functions of SiHa and HeLa cells. (a) The statistical results of optical density values in SiHa and HeLa cells overexpressing hIL-24. (b–c) Transwell experiments assessing the migration and invasion abilities of SiHa and HeLa cells overexpressing hIL-24 and (d) flow cytometry analysis evaluating the apoptosis levels in SiHa and HeLa cells overexpressing hIL-24. *P < 0.05, **P < 0.01, ***P < 0.001 compared with the empty plasmid-transfected group.

The impact of human interleukin-24 (hIL-24) knockdown on the biological functions of SiHa and HeLa cells. (a) Statistical results of optical density values in SiHa and HeLa cells transfected with the negative control (siNC) and sihIL24. (b–c) Transwell experiments assessing the migration and invasion abilities of SiHa and HeLa cells transfected with siNC and sihIL24 and (d) flow cytometry analysis measuring the apoptosis levels in SiHa and HeLa cells transfected with siNC and sihIL-24. *P < 0.05, **P < 0.01, ***P < 0.001 compared with cells transfected with siNC.
Finally, flow cytometry analysis revealed that hIL-24 significantly increased the apoptosis rate of SiHa and HeLa cells, with the apoptosis rate reaching 12.81% in the pcDNA3.1(+)-hIL-24 plasmid group (P < 0.05). Compared with the negative control group transfected with siNC, SiHa and HeLa cells transfected with sihIL-24 showed a significant decrease in the apoptosis rate (P < 0.05) (Figure 6(d), Figure 7(d)). These comprehensive data strongly suggest that hIL-24 not only inhibits the growth, migration, and invasion abilities of SiHa and HeLa cells but also promotes their apoptosis, demonstrating its tremendous potential in the treatment of cervical cancer.
Discussion
According to data from the World Health Organization on the global cancer incidence and mortality rates and global statistical analysis results from the Global Cancer Observatory database, cervical cancer ranks as the fourth most prevalent cancer in women, making it a substantial public health concern. Cervical cancer is one of the most prevalent cancers among middle-aged women in most countries.2,51 Hence, developing novel treatment strategies for cervical cancer is vital to improve the overall prognosis of patients.
The primary aim of this study was to conduct an exhaustive evaluation of the effects of hIL-24 on SiHa and HeLa cells, which are particular strains of cervical cancer. We focused on parameters involved in inhibition of cell proliferation, reduction of migration, invasion curtailment, and apoptosis induction. Moreover, research has shown that cervical adenocarcinoma accounts for approximately 20% to 25% of all malignant cervical tumors, making it the second most common cervical malignancy after squamous cell carcinoma. These two types of cancer exhibit differences in the PD-L1 immune microenvironment and tumor escape mechanisms, leading to variations in the prognostic significance and PD-L1 expression patterns among different cervical cancer types. Specifically, extensive PD-L1 expression in squamous cell carcinoma patients is associated with poorer disease-free survival and disease-specific survival, whereas in adenocarcinoma patients, tumors lacking PD-L1-positive tumor-associated macrophages demonstrate a survival advantage. 52 The knowledge gained from this study will enhance the molecular comprehension of cervical cancer and provide opportunities for novel therapeutic interventions. With cervical cancer continuing to pose a significant risk to women’s health worldwide, the development of innovative treatments is of paramount importance.53–56
The MTT assay results robustly indicated that hIL-24 markedly curtails SiHa and HeLa cell proliferation, aligning with existing research on its anti-tumor capabilities.57–60 Our findings also illuminate the signaling pathways and molecular machinations driving the anti-tumor efficacy of hIL-24, establishing a foundation for further study. Moreover, we established that hIL-24 significantly impedes the migratory and invasive potential of SiHa and HeLa cells—an important consideration for tumor metastasis management and prevention.61–63 The rigor of our experimental approach adds considerable weight to our conclusions and bolsters the case for hIL-24 as an inhibitor of metastasis. Augmenting these results, flow cytometry analysis corroborated the role of hIL-24 in promoting apoptosis in SiHa and HeLa cells, reinforcing its prospective therapeutic value.64,65
Our study’s methodological soundness and the stringent statistical tests underpinning our results lend a high degree of reliability. However, the small sample size suggests a need for further validation within a larger population.
The limitations of this study include the limited sample size and the absence of validation across multiple centers and diverse cell lines. Comprehensive research in the future should aim to rectify these limitations to fully elucidate the therapeutic promise of hIL-24 for cervical cancer.
Conclusions
Our findings clarify the role of hIL-24 in modulating the growth, migration, invasion, and apoptotic processes of SiHa and HeLa cervical cancer cells (Figure 8), enhancing our grasp of the disease’s molecular dynamics. This research provides new opportunities for clinical strategy development. Given the current lack of efficacious treatments for cervical cancer, hIL-24 may be a promising clinical therapy. However, it is crucial that subsequent studies confirm these preliminary findings and investigate the practical therapeutic applications of hIL-24 in the context of cervical cancer.
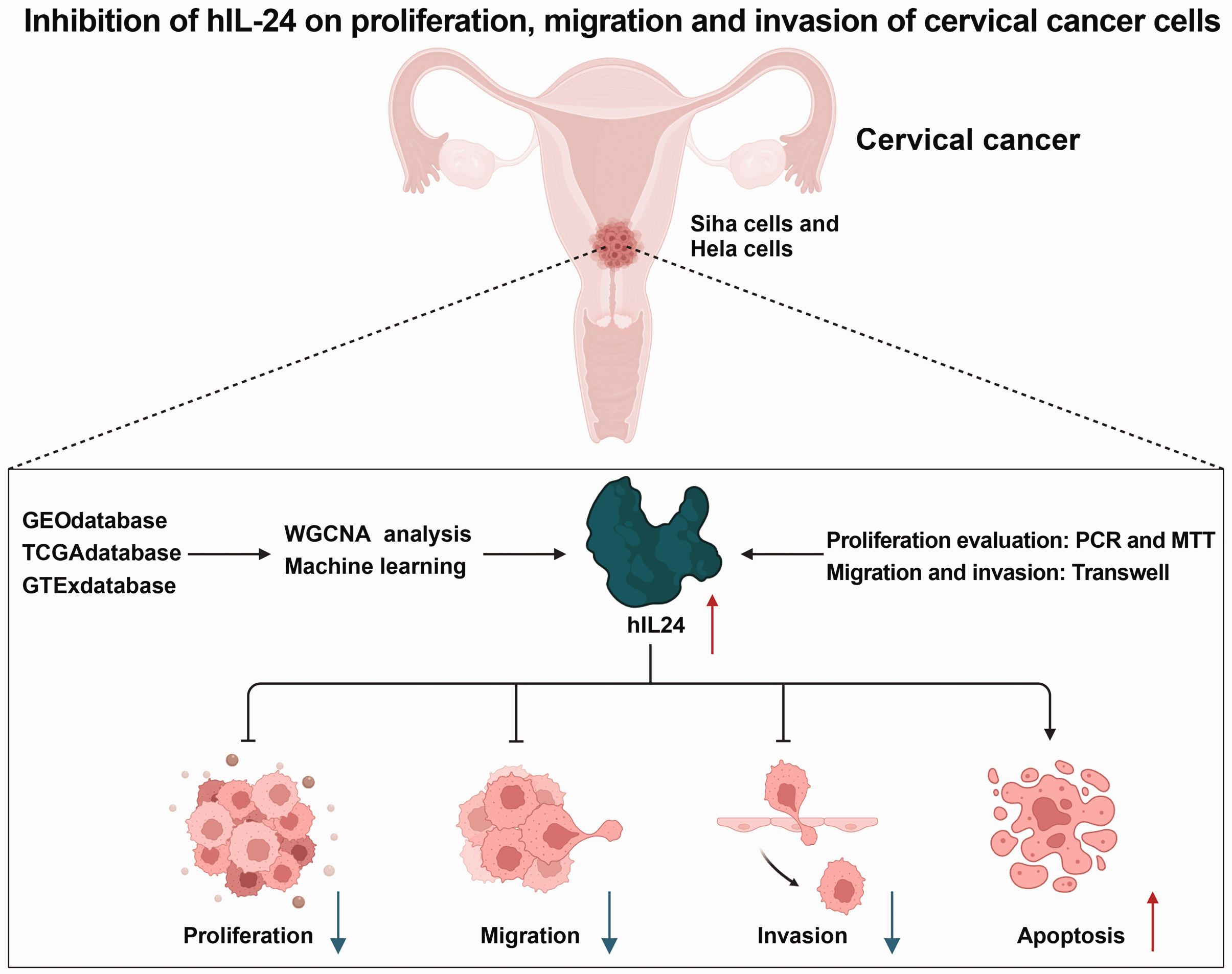
Schematic representation of the molecular mechanisms underlying the impact of human interleukin-24 (hIL-24) on the malignant biological behavior of cervical cancer cells. GEO, Gene Expression Omnibus; TCGA, The Cancer Genome Atlas; GTEx, Genotype-Tissue Expression; WGCNA, weighted gene co-expression network analysis.
Footnotes
Author contributions
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
This study was funded by the Projects of Medical and Health Technology Development Program in Shandong Province (No. 202202080776).