
Abstract
BACKGROUND
: Thyroid carcinoma is the most common endocrine malignancy worldwide. Changes in DNA methylation can cause silencing of normally active genes, especially tumour suppressor genes (TSG) or activation of normally silent genes.
OBJECTIVE
: The aim of this study is to evaluate the degree of promoter methylation for a panel of markers for thyroid neoplasms and to establish their relationship with thyroid oncogenesis.
METHODS
: To generate a comprehensive DNA methylation signature of TSGs involved in thyroid neoplasia, we use Human TSG EpiTect Methyl II Signature PCR Array-Qiagen for 24 samples (follicular adenomas and papillary thyroid carcinomas) compared with normal thyroid tissue. We extended the evaluation for three TSGs (TP73, WIF1, PDLIM4) using qMS-PCR. Statistical analysis was performed with GraphPad Prism.
RESULTS
: We noted four important genes NEUROG1, ESR1, RUNX3, MLH1, which presented methylated promoter in tumour samples compared to normal. We found new characteristic of thyroid tumours: methylation of TP73, WIF1 and PDLIM4 TSGs, which can contribute to thyroid neoplasia. A significant correlation between BRAF V600E mutation and RET/PTC rearrangements with TIMP3 and CDH13, RARB methylation, respectively was observed.
CONCLUSIONS
: TSGs promoter hypermethylation is a hallmark of cancer and a test that uses methylation quantification method is suitable for diagnosis and prognosis of thyroid cancer.
Introduction
Thyroid cancer, the most common malignant endocrine disease (more than 95% cases worldwide) [1], that can affect both genders, although it is most common in women, three to four times higher than in men. Though thyroid cancer can occur at any age, most tumours are diagnosed after the age of sixty [2].
Thyroid neoplasia represents a highly heterogeneous entity that possesses different cytological, histological and genetic characteristics. Starting with benign lesions – follicular adenoma (FA), to the well differentiated carcinomas, such as papillary thyroid carcinoma (PTC) in 85–90% of thyroid cancer cases, or follicular thyroid carcinoma (FTC) in 5–10% of all thyroid cancer cases, to poorly/undifferentiated thyroid carcinoma and anaplastic thyroid carcinoma (ATC) in 2% of thyroid cancers, or medullary carcinoma (MTC) in about 5% of thyroid cases, most primary thyroid cancers are originated from thyroid follicular cells [2].
The most prevalent type of thyroid neoplasia is papillary thyroid cancer (PTC). The survival rate is higher compared with other cancers. Prognosis for nodules less than 20 mm in diameter is excellent, but a clinically significant thyroid nodule may develop in 5% to 10% of the general population during their lifetime. 99% of patients with nodules smaller than 20 mm will be alive for 20 years, in contrast with FTC (10–20% of will be alive for 10 years), MTC (25% of patients with will be alive for 10 years) and ATC (90% of patients with will be alive for 5 years) [3].
Thyroid cancer is a multifactorial disease, the patients presenting a genetic susceptibility, but more evidences showed the involvement of epigenetic deregulated mechanisms in thyroid oncogenesis [4]. The well-known point mutations and rearrangements found in thyroid cancer tend to affect the effector genes of the MAPK pathway, such as v-raf murine sarcoma viral oncogene homolog B1 (BRAF), the RAS family and the protooncogene RET. These alterations have been shown to be subtype specific. Almost all tumours presenting RAS mutations have a follicular pattern of growth (FA, FTC or PTC follicular variant [fvPTC]). Mutations in BRAF gene and rearrangements in the RET gene are characteristic of PTC and poor prognosis [5, 6, 7]. RAS mutations have diagnostic but not prognostic value.
DNA methylation is one of the most important epigenetic modifications along with histone modifications and silencing mediated by RNA interference [8]. The epigenetic mechanisms are deregulated in cancer and the hallmark for cancer cells is represented by hypermethylation of tumour-suppressor gene and hypomethylation of repetitive DNA sequences and/or oncogenes. DNA methylation is important for transcriptional regulation, being essential for the normal cellular development. In chromosomal regions of tumour-associated genes, the epigenetic modifications can change important associated regulatory mechanisms for pathogenic malignant transformation. With DNA methylation, a methyl group is added to the fifth carbon of the cytosine residue in a CpG dinucleotide. CpG dinucleotides’ rich regions (CpG islands) are usually located in the 50-flanking gene promoter areas. Gene promoter methylation, particularly near the transcription start site, is associated with chromatin remodelling, which affects gene silencing [9, 10].
Tumour suppressor gene inactivation by promoter hypermethylation is thought to be important in carcinogenesis. Thus, measuring DNA methylation in a genome-wide manner would be valuable for studying mechanisms of epigenetic control involved in gene expression regulation. For personalized therapy, molecularly-defined tumour subgroup identification appears promising. For example, gene expression panels associated with breast cancer are now clinically used to provide individualized therapy. These panels, which depict carcinogen exposure-associated differences in individual tumours, include DNA hypermethylation involved in gene silencing as well as DNA hypomethylation, which is associated with oncogene activation and genomic instability [11, 12].
It has been observed that methylation of the thyroid cancer genes, especially methylation of tumour suppressor genes, can be correlated with clinical parameters, risk factors and the prognosis of the disease. Despite the efforts, this mechanism is not completely known.
Recent studies focused on the global methylation pattern in thyroid cancer. Ceolin et al. proved differences between the global MTC and PTC methylation level, therefore, the overall methylation profile of DNA can be correlated with thyroid cancer histology [13].
Lee et al. reported decreased levels of MAPK signal-inhibiting gene SERPINA5 in papillary thyroid cancer, due to DNA methylation [14, 15].
The aim of this study is to evaluate the degree of promoter methylation for a panel of novel and known methylation markers for thyroid neoplasms and to establish their relationship with thyroid oncogenesis.
Materials and methods
Sample collection. Biopsy samples (
All the samples were provided with patient’s agreement and the study protocol was approved by the Ethic Committee.
DNA isolation. DNA was isolated from thyroid tissue samples and blood using High Pure PCR Template Preparation Kit (Roche Molecular Biochemicals, Mannheim, Germany). DNA samples were stored at
BRAF V600E mutation was detected using PCR-RFLP analysis. PCR primer sequences used were: forward primer: 5’-GCT TGC TCT GAT AGG AAA ATG AG-3’; reverse primer: 5’-GAT AGA CAG CAG CAT CTC AGG-3’. PCR reaction mix contains: 100 ng DNA, 1.5 U/
RET split detection was performed using FISH technique. The protocol is specific for formalin-fixed paraffin-embedded (FFPE) tissue sections and consists of successive stages of dewaxing, rehydration, deproteinization and pretreatment followed by in situ hybridization with cDNA probes double labelled with fluorochromes (Zytovision GmbH – Germany). The two ends of the gene were labelled with different fluorochromes, green and orange, respectively. If the gene is intact, one see one single yellow spot. If the gene is splitted there were two distinct green and orange spots on the image. 3
RNA isolation. Isolation of total RNA was performed using Trizol technology (Invitrogen) according to manufacturer’s instructions. 50 mg of tissue was disrupted using a homogenizer and K1 cells were deposited after centrifugation and 1 ml Trizol reagent was added. Isolated RNA quality check was done by reading the concentration and purity NanoDrop spectrophotometer and Experion.
Human tumour suppressor genes DNA methylation PCR array, signature panel
The Human Tumour Suppressor Genes EpiTect Methyl II Signature PCR Array profiles the methylation status of 22 tumour suppressor gene promoters whose hypermethylation has been reported in the literature to occur frequently in a variety of cancers and tumours. Profiling tumour samples with these arrays may help correlate CpG island methylation status with biological phenotypes. The results may provide insights into the molecular mechanisms and biological pathways behind oncogenesis, as well as cancer pathology. The protocol is based on restriction enzyme digestion, followed by real-time PCR, for the analyse of the 22 different tumour suppressor genes methylation status.
Apoptosis: BRCA1, CDKN2A, DAPK1, GSTP1, MGMT (AGT), PTEN, RUNX3, TIMP3, TP73, VHL.
Cell adhesion: APC, CDH1, CDH13, CDKN2A.
Cell cycle: APC, BRCA1, CDKN2A, FHIT, MLH1, NEUROG1, PTEN, RASSF1, RUNX3, TP73, VHL.
DNA damage and repair: APC, BRCA1, MGMT (AGT), MLH1, TP73.
Signal transduction: APC, BRCA1, CDH13, ESR1, PTEN, RASSF1, SOCS1, WIF1.
Transcription factors: BRCA1, CDH1, ESR1, NEUROG1, RARB, RUNX3, TP73, VHL.
Other: PDLIM4 (RIL).
Unmethylated C residues conversion was realized with bisulphite treatment using EpiTect Bisulfite kit (Qiagen, Valencia, California, USA). 700 ng/
Primers sequence used in qMS-PCR
Primers sequence used in qMS-PCR
Primers In this purpose the promoter sequence was retrieved from EMBL database (
WIF1 gene promoter presents 3 CpG islands (199 bp, 255 bp, 357 bp) and the transcription is on the minus strand. The TSS is situated at
Direct Q-MSP of Genomic DNA is a quantitative method for evaluating the degree of sample methylation.
Standard curves were designed using positive controls (fully methylated) and negative controls (fully unmethylated). Positive and negative controls were diluted in order to obtain a serial dilution: 10 pg, 100 pg, 1 ng and 10 ng according to Absolute Quantitation Using Standard Curve Getting Started Guide (Applied Biosystems 7300/7500/7500 Fast Real-Time PCR System, Foster City, CA, USA). All DNA samples were diluted to a final concentration of 10 ng/
qPCR experiments were performed in duplicate (
Methylation percentage calculation method was done according to Fackler et al. [20]. The concentration of unmethylated and methylated DNA for each patient sample was extrapolated using the standard curves. The percentage of methylation was calculated according to the formula: % M
Cell culture treatment. In order to evaluate the role of methylation in gene expression suppression, K1 cell line culture (derived from a papillary carcinoma primary thyroid) was cultured for 24 hours in DMEM-Ham’s F12 medium supplemented with 2 mM glutamine and 10% foetal calf serum (FCS) to allow cell adherence to a minimum of 50%. Then the cells were treated with 5-azacytidine (AzaC) a DNA demethylating agent in a concentration of 5
Summary of the main clinical and pathological characteristics of samples used in this study
Apoptotic process evaluation by flow cytometry. Changes that occur in the plasma membranes are part of the early events in apoptosis. Apoptotic cells present phosphatidylserine translocated into the inner face of the plasma membrane allowing external annexin family members (proteins that bind calcium dependent phospholipids) to attach. Following the characteristic changes occurring during the apoptotic process, the test used allows a double staining: annexin V labelled with FITC (fluorescein isothiocyanate) which binds to phosphatidylserine exposed to the extracellular environment and PI (propidium iodide) – a dye used for the evaluation viability is tied intracellular DNA. For staining of cells an apoptosis kit was used (BD Bioscience, NJ, USA). More than 1
Tumour suppressor methylation percentage evaluation in tumour samples. The results were presented in descending order, according to the AUROC values
Standard curve parameters for all tested PCR primers (for methylated-M and unmethylated-U DNA sequences)
Patients clinical characteristic feature
We examined a cohort of 57 patients: 19 benign thyroid lesions (follicular adenomas) and 38 thyroid cancers (28 papillary-PTC and 10 follicular variant of papillary thyroid cancer-fvPTC). The clinicopathological characteristics and genetic tests are detailed in Table 2. The patients were grouped according to gender and the number of female patients was higher than the male. 75% from patients with thyroid tumours were positive for RET/PTC1 rearrangements, whereas 21.43% were positive for BRAF (V600E).
The human tumour suppressor genes epiTect methyl II signature PCR array
We examined 22 genes with diverse function, including cell cycle regulation, tumour suppression and DNA repair in thyroid tissues. We observed that the gene promoter methylation for each tested gene is higher in thyroid cancer tissue compared with normal adjacent tissue. The methylation for each evaluated gene is listed in Table 3. To assess the potential utility of hypermethylation of genes as molecular markers of thyroid cancer, we used ROC analysis. We evaluated the accuracy of the test measured by the area under the ROC (AUROC) curve and sensitivity/specificity. An area of 1 represents a perfect test; an area of 0.5 represents a worthless test. A rough guide for classifying the accuracy of a diagnostic test is the traditional academic point system:
0.90–1 0.80–0.90 0.70–0.80 0.60–0.70 0.50–0.60
We found that the methylation percentage was significantly higher for TP73, WIF1 and PDLIM4 gene promoter methylation. These three genes presented also the highest values for sensitivity and specificity. Good results were obtained for a panel of 8 genes (NEUROG1, RARB, ESR1, RUNX3, MLH1, RASSF1, APC, PTEN), which correlates with thyroid cancer.
The sample positive for BRAFV600E mutation presented higher methylation values for MGMT, NEUROG1, RARB gene promoters and significant methylation values for TIMP3 (Braf (
The patients positive for RET/PTC traslocation presented higher methylation values for ESR1, NEUROG1, SOCS1 gene promoters and significant methylation values for RARB (RET/PTC (
This array was realized in 24 patients on DNAs extracted from biopsy tissue (tumour vs normal). Further we evaluated the potential as biomarkers of the most important factors discovered through this type of analysis. For this purpose, in an extended patient group, we evaluated the role of WIF1, TP73 and PDLIM4 tumour suppressor genes in thyroid oncogenesis.
Validation of direct qMSP was performed for each primer pair specific amplification for methylated and unmethylated DNA in the qPCR reaction, according to our previous publications [21, 23]. The
Statistical parameters of promoter methylation status for investigated genes in patients’ samples (control vs. tumoral)
Statistical parameters of promoter methylation status for investigated genes in patients’ samples (control vs. tumoral)
The slope values were between [
The tumour samples presented significant higher methylation status than control samples (
The mRNA expression levels of WIF1, TP73 and PDLIM4 were measured in samples from thyroid carcinomas and adjacent normal thyroid tissues. The relative PDLIM4 and WIF1 mRNA expression was significantly lower (
Spearman correlation was performed to establish the influence of gene promoter methylation on gene expression. The methylation was inversely correlated with gene expression (WIF1-
The analysis of 5-azacytidine (AzaC) treatment effect upon K1 cells (concentration of 5 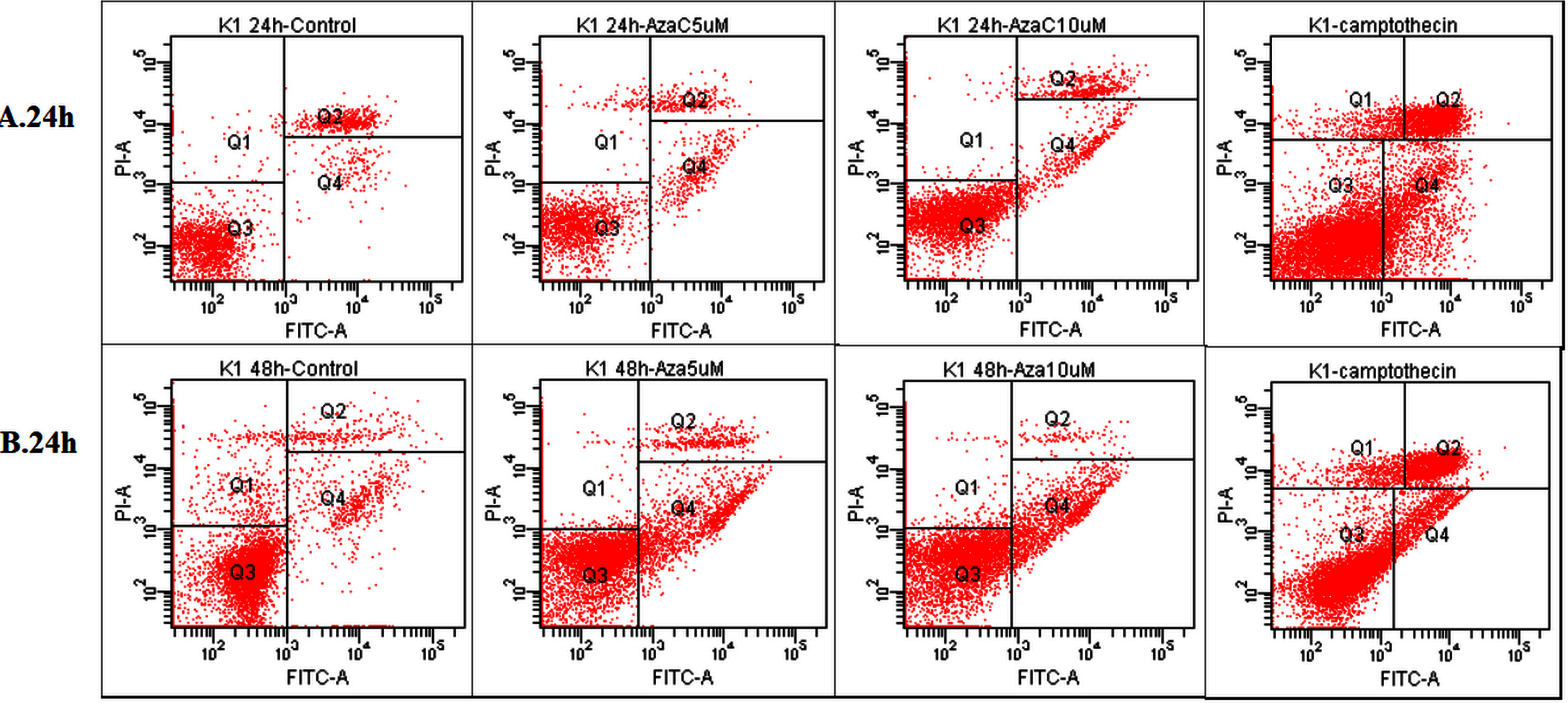
Further, we investigated the targeted gene expression and methylation promoter levels depending of the histopathological analysis.
As seen in Table 6, higher TP73 gene promoter methylation levels are characteristic for papillary carcinoma and papillary carcinoma follicular variant (
Regarding the expression of the studied genes and histopathological type statistically significant difference was observed for WIF1 gene and PDLIM4. The expression PDLIM4 and TP73 genes decrease gradually from benign neoplasia to malign status. In case of WIF1 the lowest value of gene expression was found in PTC.
Furthermore, we decided to estimate WIF1, TP73 and PDLIM4 gene promoter’s methylation as well as their expression levels in peripheral blood samples. Results revealed a high percentage of methylation for both TP73 and PDLIM4 genes in thyroid carcinoma patients, both with statistically significant difference between groups (
Regarding gene expression, it was observed that patients with thyroid carcinoma (PTC and fvPTC) exhibit reduced relative expression levels for all tested genes (Table 7).
Statistical parameters for investigated genes promoter methylation grade and n-fold genes expression of in various neoplasms for studied patients’ group
Statistical parameters for investigated genes promoter methylation grade and n-fold genes expression of in various neoplasms for studied patients’ group
Statistical parameters for methylation grade and n-fold genes expression of selected genes promoters in blood samples of studied patients’ group
The analysis results of 5-azacytidine (AzaC) treatment effect upon K1 cells
Standard K1 cells (derived from a papillary carcinoma primary thyroid) were cultured for 24 hours in DMEM-Ham’s F12 culture medium supplemented with 2 mM glutamine and 10% foetal calf serum (FBS) to allow cell adherence to a minimum of 50%. The cells were treated with 5-azacytidine (AzaC) that induces DNA demethylation in a concentration of 5
Apoptotic process evaluation by flow cytometry
The untreated cell population was used as control to define the basal level of apoptotic and dead cells, while the cells incubated for 3 hr at 37
As shown in Fig. 1 the treatment with 5-azacytidine of K1 tumour cell line reduces the cell viability (Q3) in a time and concentration depending manner.
The cell viability is reduced as a response to 5-azacytidine treatment correlated with an increase in K1 cells apoptotic process Table 8.
As shown in the Fig. 1, the treatment of K1 tumour cell line with 5-azacytidine determines an increase of both the early and late apoptosis, also depending on time and concentration.
5-azacytidine induces an increase of early apoptosis 2.5 times (9.8%) compared with untreated cells (3.9%), after 24 hours of exposure at 5
The data showed that 5-azacytidine, a DNA deme- thylating agent enhanced the apoptotic process of K1 cell line, suggesting a possible link between the DNA
Effect of 5-azacytidine treatment on the target genes methylated promoters.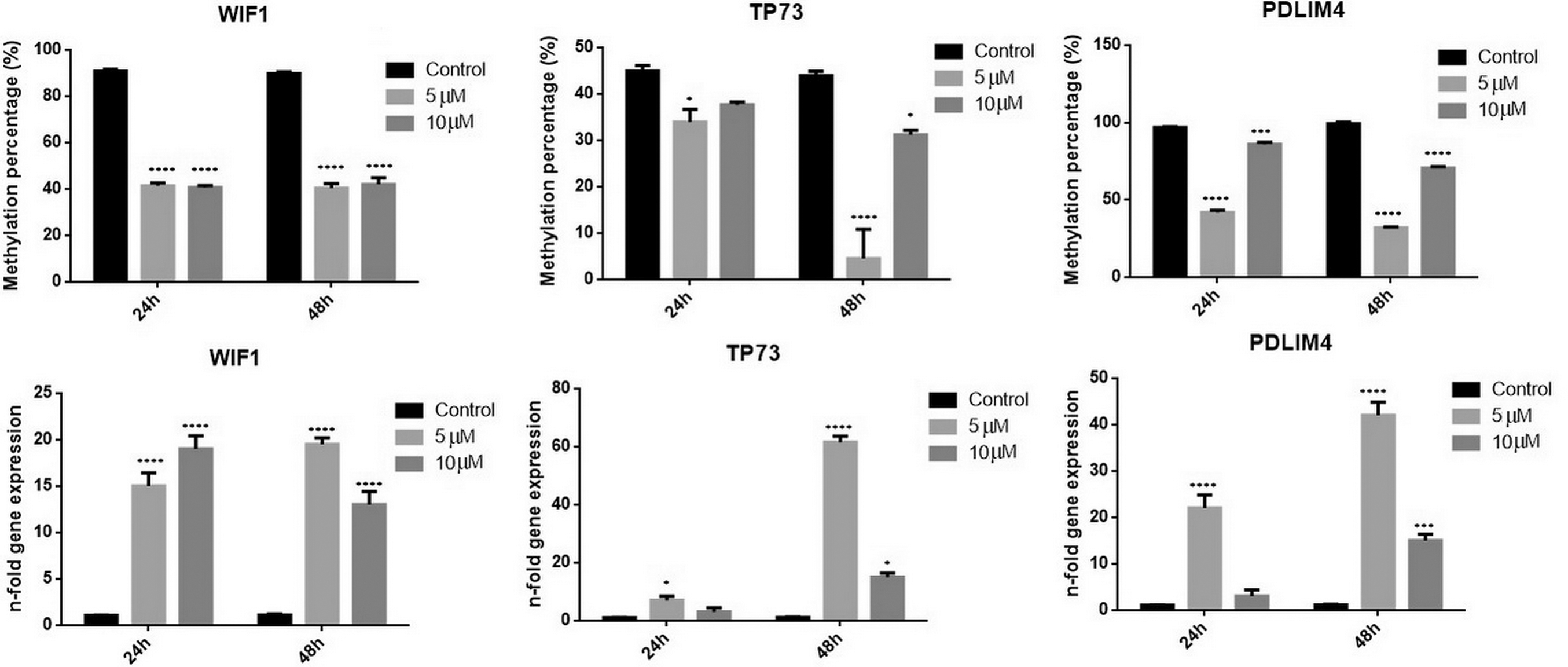
methylation status and the activation of apoptotic pathways.
Further, we evaluated the effect of 5-azacytidine upon the promoter of target genes. As shown in the Fig. 2 the promoter methylation percentage decreases considerably for all three genes. The promoter is demethylated especially after a 5
Consecutive, the gene expression increases significantly, the promoter becoming active. The highest increment was observed for TP73 gene (
Discussion
Along with various molecular alterations found in thyroid neoplasia (mutations and/or gene rearrangements) that were described (RET germline mutation, and chromosomal rearrangements resulting in the fusion gene PAX8/PPAR
Like other human tumours, aberrant methylation of tumour suppressor genes is also a characteristic for thyroid tumours. Some reports mention the involvement in thyroid tumorigenesis of aberrant silencing trough promoter methylation of PTEN [25], RASSF1A [26, 27], tissue inhibitor of metalloprotei- nase-3 (TIMP3), SLC5 A8, death-associated protein kinase (DAPK) and retinoic acid receptor
Our study confirms the precedent reports regarding the gene promoter methylation of RARB, RASSF1, PTEN in thyroid cancer. According to gene panel studied we found new factors characteristic to thyroid tumours: methylation of TP73, WIF1 and PDLIM4 tumour suppressor genes, which can be a contributor to thyroid neoplasia.
The TP73 gene is a member of TP53 family involved in controlling cellular proliferation, differentiation and cell death, which includes also the TP63 gene [30]. TP73 role compensates for the loss of p53 function, and is rarely mutated compared to TP53 [31]. One interesting fact is that TP73 gene transcription is controlled by two different promoters (P1 and P2), leading to p73 protein isoforms with opposite functions, one containing (TA) or lacking (
Hypermethylation of the TP73 gene promoter was also found in a variety of solid tumours such as squamous cell lung cancer [35], gastric carcinoma [36] and cervical cancer [37].
The current study showed that the methylation of TP73 gene promoter is significantly higher in tumour tissue compared to normal tissue (
The WNT inhibitory factor-1 (WIF-1) gene is a secreted antagonist that binds to WNT proteins to suppress WNT/
WIF-1 gene expression was found to be down-regulated through promoter hypermethylation, geno- mic loss, or genomic rearrangement in many human neoplasia, including lung, colon, breast, bladder, kidney, prostate and salivary gland tumours [40, 41, 42, 43, 44, 45, 46].
We showed that WIF1 is hypermethylated in tumour samples, the percentage of methylation being significantly higher (
PDLIM4 (reversion-induced LIM gene-RIL) was a potential tumour suppressor gene involved in cell growth regulation and modulates actin stress fibre dynamics through its association with alpha-actin [47]. Moreover, it has been shown that PDLIM4 promoter methylation could be the main mechanism to suppress its expression in prostate cancer tissues [48]. Other report showed also an association of PDLIM4 promoter methylation with breast cancer [49]. In colon cancer hypermethylation of PDLIM4 suppresses activation of Src (a regulatory protein that functions in several fundamental processes, including cell differentiation, proliferation, migration, and survival) [50]. PDLIM4 promoter is hypermethylated in tumour samples, especially in PTC samples. Follicular adenomas presented higher PDLIM4 methylation levels, suggesting the possibility to use this factor as a marker for diagnosis as long as significant methylation levels were quantified from blood of patients with thyroid neoplasms. Lowest PDLIM4 expression values were found in fvPTC samples.
Detection of methylation levels of the tested genes in blood could represent a potential method to evaluate the patient status. Even if PDLIM4 and TP73 genes expression decreases gradually from benign neoplasia to malign status, and WIF1 presented the lowest value in PTC samples; evaluation of gene expression in blood does not provides the same informational value.
We indicated also that the treatment with demethylating agents as 5-AzaC could restore the gene function as the expression increases significantly and the promoter becomes active. The increment was notable especially for TP73 gene, but also for PDLIM4 and WIF1 gene.
Among the tested genes we noted four more important genes NEUROG1, ESR1, RUNX3, MLH1, which presented the promoter with higher methylation values in tumour samples compared to normal.
The genetic tests for mutations in thyroid cancer are innapropiate due to the low incidence of mutations in thyroid oncogenesis [51] and no specifc inactivating mutations have been identifed in well-differentiated thyroid cancers [52, 53, 54]. However, BRAF V600E mutation is present in papillary thyroid cancer and is associated with an aggressive tumor phenotype and higher risk of recurrent and persistent disease [55].
Various studies suggested the involvement of BRAF V660E mutation in aberrant methylation processes of certain genes in colon cancer [56, 57] and in thyroid cancer [10, 29]. A high-throughput study demonstrated the effect of BRAF V600E signalling on aberrant methylation that may have a profound impact on cell functions and behaviors of PTC [58]. We found also a significant correlation between Braf V600E mutation presence and TIMP3 gene promoter methylation, but also with MGMT, NEUROG1, RAR-beta gene promoter methylation.
RET/PTC rearrangements were associated with a more aggressive phenotype, a larger tumor size, and it was shown to influence the methylome [59, 60]. The present study showed a significant correlation between RET/PTC rearrangement and CDH13, RARB genes promoter methylation. A correlation was also observed for this translocation with ESR1, NEUROG1 and SOCS1 gene promoter methylation.
Conclusions
The hypermethylation of tumour suppressor promoter is a hallmark of cancer and by inhibiting the genes expression, it alters the cell signalling pathways. Therefore, a test that uses as a method for methylation quantification of tumour suppressor genes is suitable for diagnosis and prognosis of thyroid cancer.
Footnotes
Acknowledgments
This research was funded by national partnership research project: PNII-PCCA 135/2012. The study was also supported by Project 433/ID 929/SMIS code 14049, SOP-IEC, Operation 2.2.1, POSCCE 2.2.1, ID 918, SMIS 14045.
Conflict of interest
The authors declare no conflict of interest.
Authors’ contributions
All the authors contributed equally.