
Abstract
BACKGROUND:
Aberrant epigenetic patterns are a hallmark of acute myeloid leukemia (AML). Mutations in profound epigenetic regulators DNMT3A and IDH1/2 often occur concurrently in AML.
OBJECTIVES:
The aim was to analyze DNA methylation, hydroxymethylation and mRNA expression profiles in AML with mutations in DNMT3A and IDH1/2 (individually and in combinations).
METHODS:
Infinium MethylationEPIC BeadChip (Illumina) covering 850,000 CpGs was utilized. The validation of hydroxy-/methylation data was done by pyrosequencing. HumanHT-12 v4 Expression BeadChip (Illumina) was used for expression examination.
RESULTS:
Hierarchical clustering analysis of DNA hydroxy-/methylation data revealed clusters corresponding to DNMT3A and IDH1/2 mutations and CD34+ healthy controls. Samples with concurrent presence of DNMT3A and IDH1/2 mutations displayed mixed DNA hydroxy-/methylation profile with preferential clustering to healthy controls. Numbers and levels of DNA hydroxymethylation were low. Uniformly hypermethylated loci in AML patients with IDH1/2 mutations were enriched for immune response and apoptosis related genes, among which hypermethylation of granzyme B (GZMB) was found to be associated with inferior overall survival of AML patients (
CONCLUSIONS:
Distinct molecular background results in specific DNA hydroxy-/methylation profiles in AML. Site-specific DNA hydroxymethylation changes are much less frequent in AML pathogenesis compared to DNA methylation. Methylation levels of enhancer located upstream GZMB gene might contribute to AML prognostication models.
Introduction
Acute myeloid leukemia (AML) is a predominantly fatal hematological malignancy and displays great molecular heterogeneity. Numerous genomic next-generation sequencing based studies have greatly contributed to a more comprehensive understanding of AML pathogenesis. However, AML exhibits less
Characteristics of AML samples examined by DNA hydroxy-/methylation and gene expression profiling
Characteristics of AML samples examined by DNA hydroxy-/methylation and gene expression profiling
Variant allele frequency (VAF) and position of mutations present in selected acute myeloid leukemia (AML) diagnostic samples is shown. AML patients chosen for this study were selected from 258 consecutive non-APL intensively treated AML patients that were examined for their mutational status by ClearSeq AML kit (Agilent Technologies, USA) in the concurrent study [Folta et al., Br. J. Haematol. 2019, doi: 10.1111/bjh.15916, in press]. F – female, M – male, VAF – variant allele frequency.
genetic lesions when compared to most solid tumors [1]. Presumably other factors, such as epigenetic changes, may significantly contribute to initiation and progression of AML. Indeed, mutations of genes involved in DNA methylation and hydroxymethylation pathways are found in approx. 40% of AML patients at diagnosis [1]. Here, we focused on mutations in DNA methyltransferase 3A gene (DNMT3A
Patients
This study conforms with The Code of Ethics of the World Medical Association, it was approved by the Institutional Ethics Committee and all patients provided their full consent. Twenty-four de novo cytogenetically normal (CN) AML patients selected from the concurrent collaborative mutational study [Folta et al., Br. J. Haematol. 2019, doi: 10.1111/bjh.15916, in press; for more details see a legend of Table 1] were divided according to their mutational status into 5 groups: DNMT3A
Sample preparation
DNA and RNA from peripheral blood leukocytes were extracted using AllPrep DNA/RNA Mini Kit (Qiagen, Hilden, Germany). Mononuclear cells of healthy blood donors were harvested from buffy coats by Ficoll gradient centrifugation (Histopaque, Sigma-Aldrich). CD34+ cells were then isolated using MicroBeads kits (Miltenyi Biotec, Bergish Gladbach, Germany) and DNA was extracted using MagCore system (RBCBioscience, New Taipei City, Taiwan).
Methylation and hydroxymethylation assessment
DNA (1.2
Pyrosequencing
BS and oxBS converted DNA (10–20 ng) was amplified using PyroMark PCR kit (Qiagen). For primer sequences see Table S1. Pyrosequencing was performed on PyroMark Q24 instrument (Qiagen).
Gene expression analysis
Expression profiles of all AML samples and CD34+ cells from healthy controls (
Summary of methylation and hydroxymethylation results
Summary of methylation and hydroxymethylation results
Hierarchical clustering of samples based on DNA hydroxy-/methylation and gene expression data. Hierarchical clustering of DNA hydroxy-/methylation and gene expression was performed by pvclust package [14] using multiscale bootstrap resampling (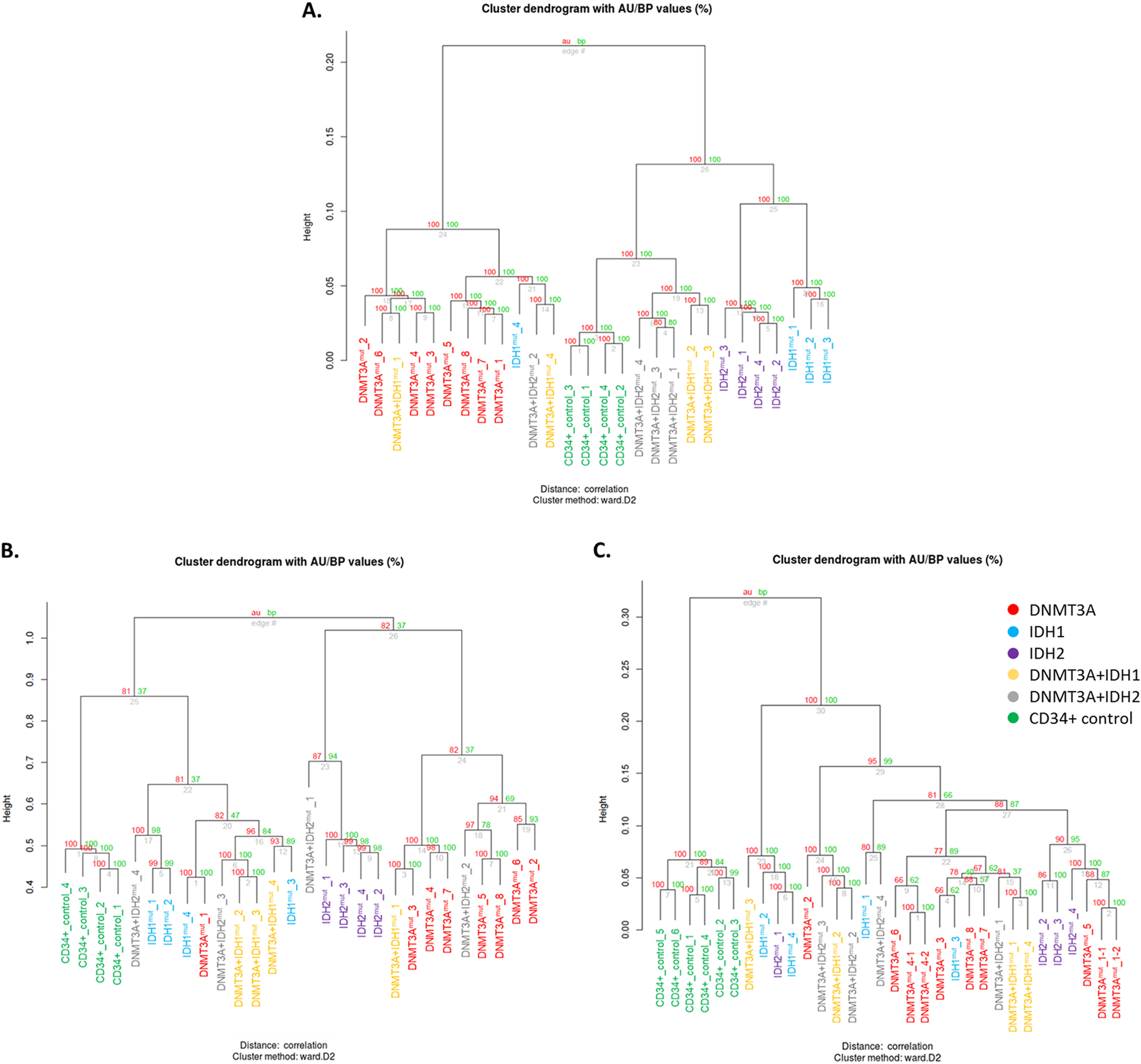
Kaplan-Meier curves and two-sided log-rank test were used to estimate the overall survival (OS) and event-free survival (EFS) using GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA) at a level of significance of 0.05. Cox regression analysis was performed in the SPSS software (IBM, Armonk, NY, USA). Validation of hydroxy-/methylation data and correlation of DNA methylation and expression datasets was assessed based on Pearson correlation coefficient (R) and F-test of overall significance.
Results and discussion
Analysis of EPIC array data
DNA methylation
After quality filtering steps, 716,847 probes for each sample remained for a subsequent analysis. Hierarchical clustering revealed 3 main DNA methylation clusters (Fig. 1A) – DNMT3A
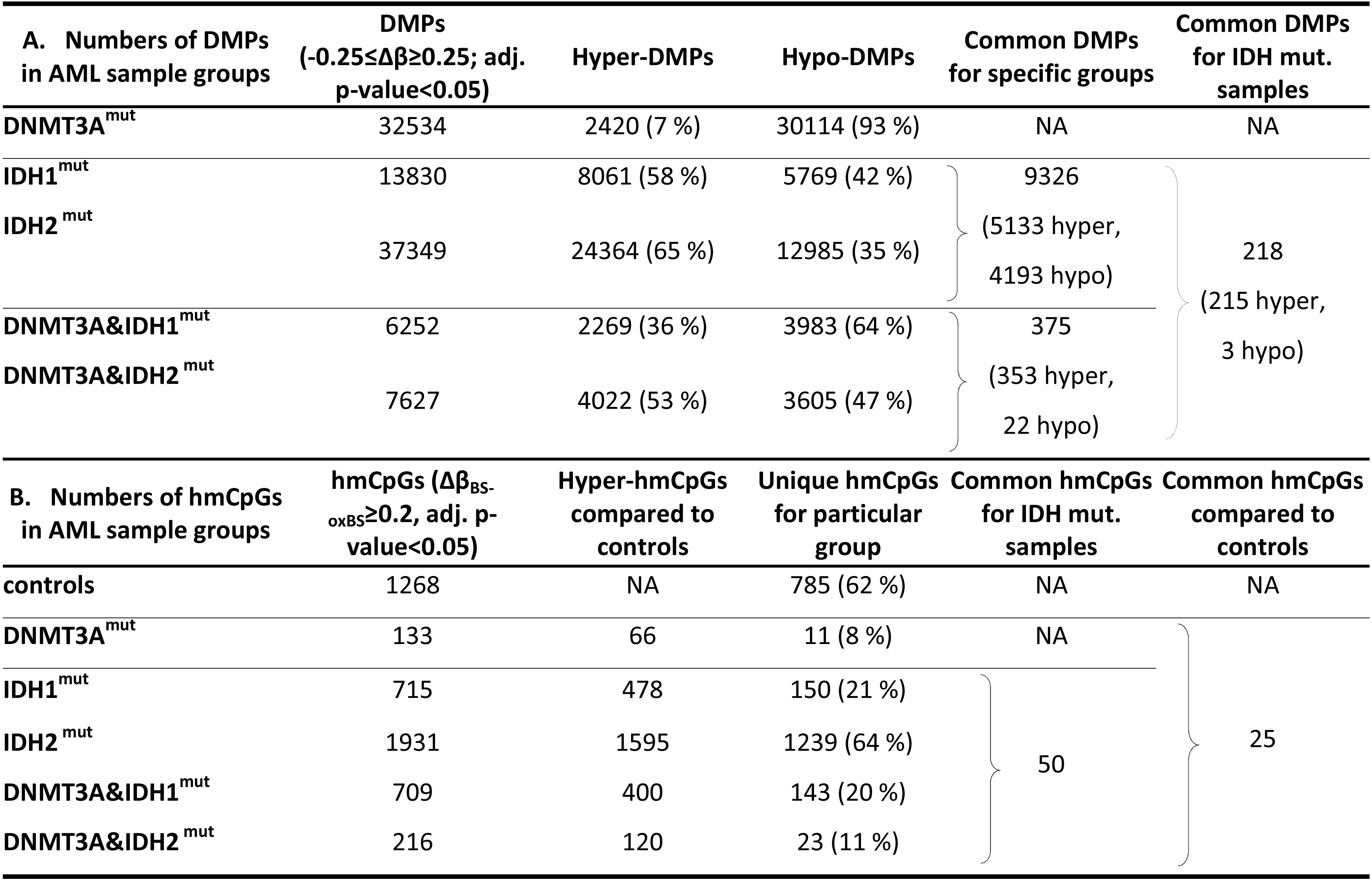
Numbers of differentially methylated positions (DMPs) are depicted in Table 2A. We observed a profound hypomethylation in DNMT3A
DNA hydroxymethylation
A rather conservative threshold of
Validation of DNA methylation and hydroxymethylation data
BS-pyrosequencing was utilized for DNA methylation validation of GZMB (cg00771752) and CHFR gene (cg00338702). For both examined loci, the Pearson correlation coefficient (
OxBS-pyrosequencing of 2 selected loci of MYB (cg23073132) and RNF216 (cg05617317) did not completely reproduce hydroxymethylation results obtained from EPIC array.
Influence of comutations (other than DNMT3A and IDH1/2) on DNA hydroxy-/methylation profile
Despite our effort to select molecularly homogenous diagnostic AML cohort, certain degree of heterogeneity could not be avoided due to the nature of AML often presenting more mutations simultaneously. In our AML mutational subgroups, NPM1 mutations were the most often occurring comutation (15/24 AML). Interestingly, only one of IDH1/2
Regarding DNA hydroxymethylation, presence of TET2 mutation in two DNMT3A
GZMB methylation levels in connection with AML prognosis. A. Overall survival of AML patients based on GZMB methylation levels. B. Event-free survival of AML patients based on GZMB methylation levels.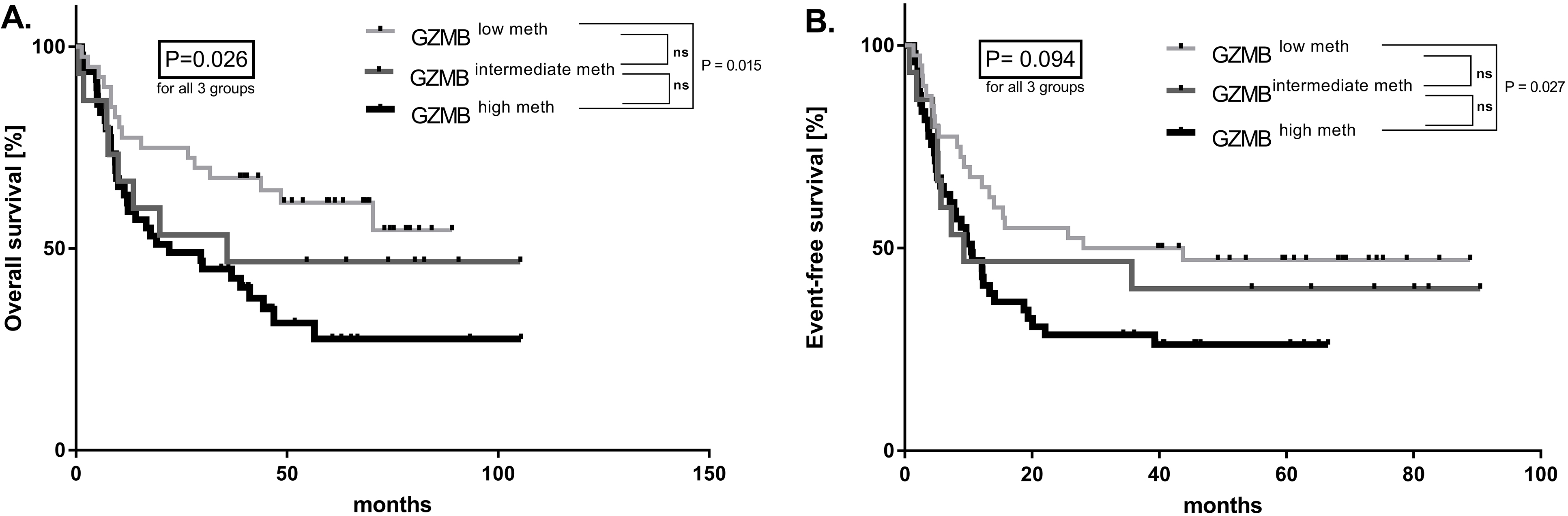
Hierarchical clustering of 10,067 probes (Table S4) with detection
Evaluation of GZMB and CHFR methylation levels as a potential prognostic biomarkers
The human granzyme B protein (GrB) encoded by GZMB gene plays a crucial role in induction of apoptosis [2]. We measured DNA methylation of GZMB enhancer (located approx. 40 kb upstream) covering two adjacent CpG sites in 104 diagnostic AML patients (for patients characteristics see Table S5). Healthy donors samples (
Multivariate Cox regression analysis of prognostic factors in AML patients for overall and event-free survival
Multivariate Cox regression analysis of prognostic factors in AML patients for overall and event-free survival
Prognostic variables and corresponding hazard ratios (HR), confidence intervals (CI) and
In this study, we found out that specific mutational background in AML patients is connected with distinct DNA hydroxy-/methylation patterns based on presence of DNMT3A
Footnotes
Acknowledgments
This work was supported by Ministry of Health of the Czech Republic, grant no. 15-25809A, all rights reserved, and by the project (Ministry of Health, Czech Republic) for conceptual development of research organization (00023736, IHBT). Access to computing and storage facilities owned by parties and projects contributing to the National Grid Infrastructure MetaCentrum provided under the programme “Projects of Large Research, Development, and Innovations Infrastructures” (CESNET LM2015042), is greatly appreciated.
Conflict of interest
The authors declare that they have no competing interest.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-182176.