
Abstract
BACKGROUND:
Liver carcinoma is a major cause of cancer-related death worldwide. Up to date, the mechanisms of liver cancerigenesis and development have not been fully understood. Multi-genes and pathways were involved in the tumorigenesis of liver cancer.
OBJECTIVE:
The aim of the present study was to screen key genes and pathways in liver cancerigenesis and development by using bioinformatics methods.
METHODS:
A dataset GSE64041 were retrieved from GEO database and the differentially expressed genes (DEGs) were screened out. Then the DEG functions were annotated by gene ontology (GO) and pathway enrichment analysis, respectively. The hub genes were further selected by protein-protein interaction (PPI) analysis. Afterwards, the mRNA and protein expressions as well as the prognostic values of the hub genes were assessed.
RESULTS:
As a result, 208 up-regulated and 82 down-regulated genes were screened out. These DEGs were mainly enriched in cell cycle and metabolism-related pathways. Through PPI analysis, TOP2A, PRDM10, CDK1, AURKA, BUB1, PLK1, CDKN3, NCAPG, BUB1B and CCNA2 were selected as hub genes, which were all over-expressed in liver cancers relative to those in normal tissues, respectively. Among them, PLK1 and CCNA2 were suggested to be prognostic factors for liver carcinoma.
CONCLUSION:
In conclusion, the present study identified several hub genes, and cell cycle and metabolism-related pathways that may play critical roles in the tumorigenesis of liver cancer. Future validation laboratory experiments are required to confirm the results.
Introduction
Liver cancer, usually known as hepatocellular carcinoma (HCC), accounts for 70–85% primary malignant tumor in the liver, which is a major cause of cancer death and its incidence is increasing [1]. Previous reports have indicated that several epidemiological factors such as aflatoxins exposure [2], chronic virus hepatitis [3], and genetic factors [4] might have an association with liver cancer risk. Nevertheless, the underlying mechanisms of liver cancer genesis and development are still unclear.
The genesis of liver cancer is a complex biological process involving multiple genes and steps. Evidence suggested that dysfunctional angiogenesis, chronic inflammation, interaction of proinflammatory cytokines, endocrine hormones, adipokines and cell metabolism alteration might be involved in cancer development and progression [5]. Thus, the treatment strategy for liver cancer has been comprehensive therapy such as surgery, chemotherapy, and even biotherapy with differing advantages and disadvantages [6]. Nevertheless, most patients with liver cancer are diagnosed at intermediate or advanced disease stages, and in this case curative approaches are often not feasible. The current standard treatment for patients with advanced liver cancer is the multikinase inhibitor Sorafenib [7]. Though Sorafenib can significantly increase overall survival in these patients, seven large, randomized phase III clinical trials evaluating other molecular therapies in the first-line and second-line settings have failed to improve on the results observed with this drug [8]. Therefore, effective treatment options are needed for advanced liver cancer patients.
The exact mechanisms of liver cancerigenesis are still unknown. A number of studies have revealed possible roles of some genes and pathways such as PIN1 [9], Wnt/beta-actin [10], and Nrf2/Keap1 [11] in the development of liver cancer. However, these reports only concentrated on any certain molecule, gene or pathway, ignoring that the cancerigenesis and development process involves aberrant expression of a variety of genes and pathways, among which some hub genes/proteins might diffusively interact with other genes/proteins and thus play a key role in the malignant transformation process of cells [12]. Moreover, these hub genes/proteins may act as prognostic biomarkers or treatment targets for cancers [13]. Therefore, it is urgent to explore novel biomarkers for liver carcinoma with a powerful genome-wide technology.
Microarray is one of the widely used high-through- put tool for performing global gene expression profiles. In the present study, we aimed to find possible hub genes/proteins and screen prognostic biomarkers for liver carcinoma by using bioinformatics methods. The differentially expressed genes (DEGs) between liver cancer tissues and paired non-cancerous tissues were screened out by analyzing the microarray-based big data. Then, the functions and roles of selected candidate genes in liver cancer were further evaluated. A protein-protein interaction (PPI) network was constructed to screen the hub nodes (hub genes/proteins) that have been found to play important roles in many networks, because highly connected hub genes are expected to play critical roles in biological processes [14]. Afterwards, both the mRNA and protein expressions as well as the prognostic values of the hub genes were assessed.
Materials and methods
Data source
To obtain microarray-based big data, datasets were retrieved in the Gene Expression Omnibus (GEO) database (
Datasets met the following criteria were considered. First, the study type must be Expression profiling by array; second, the studies should only concern human sapiens; third, the study must include both liver cancer tissues and paired non-cancerous tissues. As a consequence, a gene expression profile (GSE64041) comprising 60 liver carcinoma tissue samples and 60 paired non-cancerous liver specimens, which was deposited by Makowska et al. [15], met the criteria, and thus it was selected for further analysis.
Screening of DEGs
To gain the DEGs between liver cancer and non-cancerous tissue, the data of GSE64041 were submitted to GEO2R that is based on limma R packages from the Bioconductor project [16] for comparison. The data were downloaded for further screening. Genes that met the cut-off criteria of
Functional enrichment analysis of DEGS
To learn the possible functions of the DEGs, the functional enrichment analyses of the DEGs, mainly involving gene ontology (GO) function analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were carried out by using Gather tool [17]. In the GO analysis, genes were classified into hierarchical categories and the gene network was presented according to biological processes [18]. A
The most significant up-regulated and down-regulated DEGs in GSE64041 (Top ten, liver cancer versus non-cancerous tissue)
The most significant up-regulated and down-regulated DEGs in GSE64041 (Top ten, liver cancer versus non-cancerous tissue)
To screen the possible hub genes/proteins that might play a central role in the development of liver cancer, the DEGs were submitted to STRING and Cytoscape tools to predict the interaction relationship among them. In the STRING tool, a combined score of not less than 0.40 (median confidence score) was considered significant. The hub proteins were selected based on its relations with other proteins, which were sorted by the degree value in the network.
Assessment of the mRNA expression of the hub genes
To evaluate the comparison of the mRNA expression levels of the selected hub genes in liver cancer vs normal tissues, the relevant data were assessed in Oncomine database that is a platform containing about 18,000 cancer gene expression microarrays for researchers [20]. The individual gene expression level of the hub genes was analyzed in this database. The rank for a gene is the median rank for that gene across each of the analyses. For the comparisons, 1.5 fold change, P-value
Assessment of the protein expression of the hub genes
To further learn the protein expression of the hub genes in liver cancer and normal liver tissues, they were respectively evaluated in the Human Protein Atlas (HPA) database [21]. The proteins were assessed by immunohistochemistry in tissue microarrays. The staining/expression of the proteins were classified according to the depth of stain as not detected, low, medium and high.
Assessment of the prognostic value of the hub genes
To evaluate the prognostic value of the selected hub genes in liver carcinoma, the top 10 hub genes were submitted to ProggeneV2 database [22] for analysis. A liver cancer cohort in The Cancer Genome Atlas (TCGA) was used to assess the effect of the genes/proteins on the overall survival time of the patients. The probability of survival and its significance was assessed by using Kaplan-Meier method and log-rank test, respectively. Moreover, hazard ratio (HR) value was also calculated. Two-tailed P value of less than 0.05 was considered as statistically significant.
Go analysis (a) and KEGG enrichment analysis (b).
Identification of DEGs from the selected gene expression profile
The dataset GSE64041 was retrieved from GEO database, which was a gene expression profile conducted on Affymetrix Human Gene 1.0 ST Array platform. GSE64041 included 60 cancer specimens and 60 paired non-cancer specimens. After the comparison, a total of 208 up-regulated and 82 down-regulated genes were screened out according to the criteria (Table 1, Supplementary Table 1).
Functional annotation and pathway enrichment of DEGS
To perform the function annotation of the DEGs, GO analysis was conducted. The results showed that the DEGs were enriched in 68 GO items, most of which were enriched in items regarding cell cycle and cell metabolism, such as GO:0000278 [6]: mitotic cell cycle, GO:0000280 [7]: nuclear division, GO:00454 49 [6]: regulation of transcription, GO:0007088 [7]: regulation of mitosis, and GO:0019222 [4]: regulation of metabolism. The top 20 GO items were listed in Fig. 1a.
Protein-Protein Interaction networks of the 10 hub genes (proteins) including TOP2A, PRDM10, CDK1, AURKA, BUB1, PLK1, CDKN3, NCAPG, BUB1B and CCNA2.
The hub genes in the protein-protein interaction network (top 10)
Pooled analyses on the mRNA expression of the ten hub genes in liver carcinoma vs normal tissues.
The results of KEGG pathway analysis showed that the DEGs were enriched in 6 pathways, including gamma-Hexachlorocyclohexane degradation, Tryptophan metabolism, Fatty acid metabolism, Bile acid biosynthesis, Cell cycle, and Stilbene, coumarine and lignin biosynthesis, in accordance with the results of the GO analysis (Fig. 1b).
In order to screen the hub genes/proteins, the DEGs were submitted to STRING website for analysis. The DEGs with a confidence of 0.40 were involved for establishing PPI networks. In the network, the top 10 node proteins, including TOP2A, PRDM10, CDK1, AURKA, BUB1, PLK1, CDKN3, NCAPG, BUB1B and CCNA2, showed a strong association with other proteins (more than 5), suggesting that they have higher hub degrees (Table 2 and Fig. 2). Therefore, the hub genes/proteins may play a central role in the genesis and progression of liver carcinoma.
Over-expression of mRNA of the hub genes in liver carcinoma
The mRNA expression levels of TOP2A, PRDM10, CDK1, AURKA, BUB1, PLK1, CDKN3, NCAPG, BUB1B and CCNA2 were analyzed in Oncomine database, respectively. The clinical specimens of liver cancer vs normal tissue were compared, of which the data met the inclusion criteria (P value of less than 0.05) were involved. There are five datasets involved in this analysis, as shown in Fig. 3. For each gene, a comparative combined analysis was conducted and an average P value was presented. The P values for the ten hub genes were all less than 0.05 and the columns were red or white, but not blue, indicating that the mRNAs of the ten hub genes were significantly up-regulated in liver cancer tissues, compared with those in normal tissues.
Assessment of the protein expression levels of TOP2A, PRDM10, CDK1, AURKA, PLK1, NCAPG, and CCNA2 in liver carcinoma and normal tissues.
To evaluate the protein expression of the hub genes in liver carcinoma, relevant data were retrieved from the HPA database. As a consequence, expression of 7 out of 10 hub proteins both in liver cancer and normal liver tissue were available in the database. There was a lack of relevant data regarding the remaining 3 proteins, namely, BUB1, BUB1B and CDKN3, in the database. As shown in Fig. 4, ‘medium’ to ‘high staining’ of most hub proteins were shown in liver cancer, while ‘not detected’ to ‘low staining’ in normal tissues, indicating that these hub proteins were over-expressed in liver carcinoma, compared with those in normal tissues.
The prognostic value of the hub genes
To evaluate the prognostic values of the hub genes, the top 10 hub genes were assessed in a TCGA liver cancer cohort, respectively. The gene expressions were divided into two groups (high expression and low expression) according to the median expression level. The survival curves were drawn to assess the relationship between the hub gene expression levels and the prognosis of liver cancer, respectively. As a result, among the ten hub genes, expressions of PLK1 and CCNA2 might affect the overall survival time of liver cancer patients. The log-rank test showed that the patients with high expression of PLK1 [HR: 1.15 (1.01–1.31),
The overall survival Kaplan-Meier curves in liver cancer patients with high and low expression of PLK1 (a) and CCNA2 (b). The P value of the log-rank test for PLK1 was 0.040 (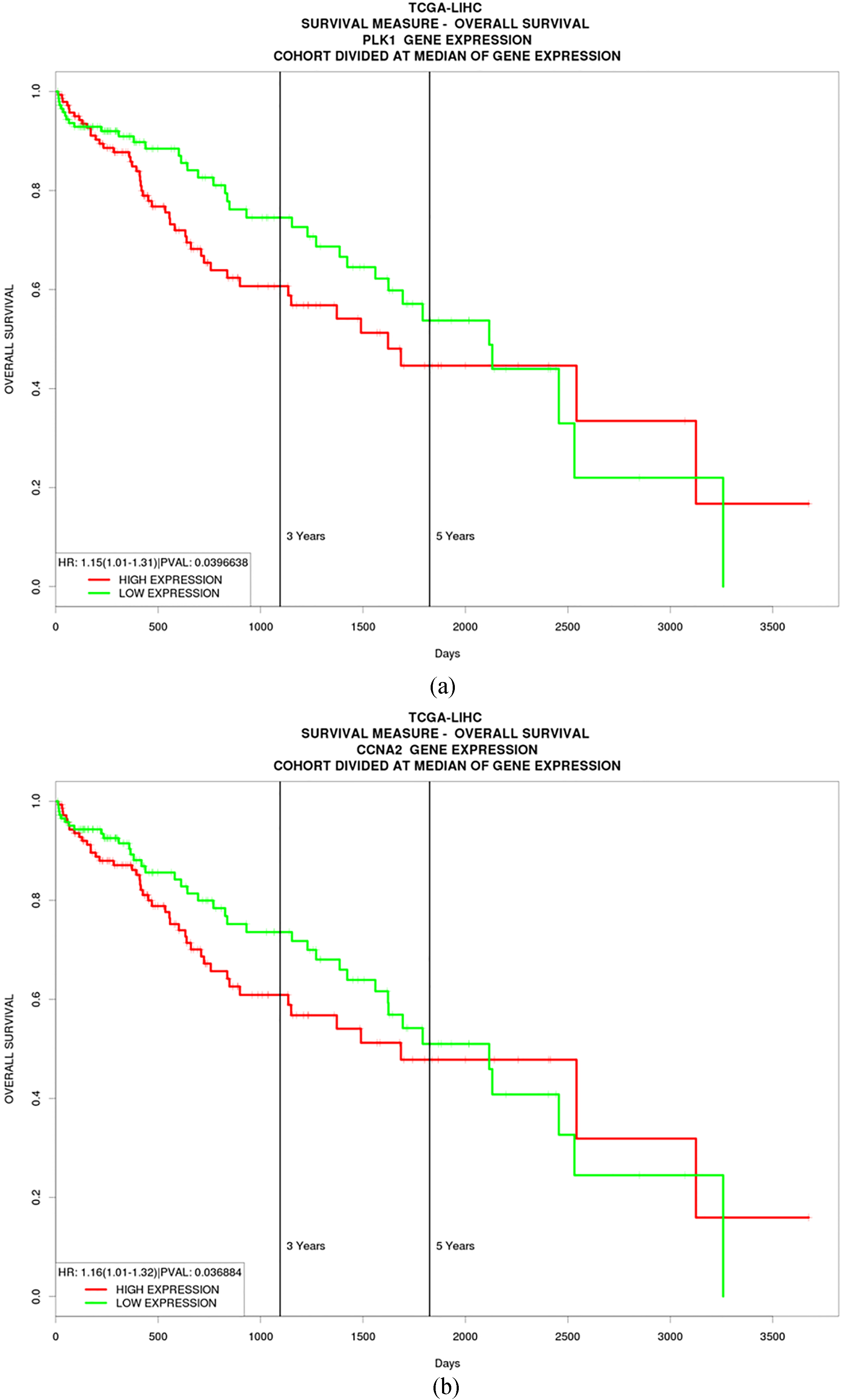
In the present study, a gene expression profile GSE64041 containing 60 liver cancer specimens and paired non-cancerous specimens were retrieved and analyzed. DEGs were obtained and their functions were annotated by GO and KEGG pathway analysis. Most of the DEGs were enriched in pathways regarding cell cycle and cell metabolism. Several hub genes were screened out, including TOP2A, PRDM10, CDK1, AURKA, BUB1, PLK1, CDKN3, NCAPG, BUB1B and CCNA2, of which PLK1 and CCNA2 might have a prognostic value for liver carcinoma.
The genesis and development of liver carcinoma is a complicated biological process that includes multi-steps and genes. Previous studies have addressed the molecular mechanisms of liver carcinoma via bioinformatics methods. In 2015, Liu et al. [23] screened DEGs of liver cancer vs normal controls by pooling GSE6222 and GSE41804, and found several key genes. However, GSE6222 contained only 6 liver cancer samples and 2 healthy control samples, while GSE41804 included 10 liver cancer samples and 10 non-cancerous samples. The sample sizes of these two datasets were limited. Besides, the samples in GSE41804 were related to both HCV infection and Il28B genetic variation, indicating that the samples of GSE41804 could not be representative of a great proportion of liver carcinoma. In the present study, 60 liver cancer specimens and 60 paired non-cancer specimens were involved. The larger sample size and the paired controls might increase power to address the mechanisms of liver cancer genesis. Recently, another study conducted by He et al. [24] compared the liver cancer tissues and cirrhosis, and screened out the DEGs. Then, the functions of DEGs were further annotated. However, though cirrhosis is regarded as a pre-cancerous stage of liver cancer, not all liver cancer cases were developed from cirrhosis. Hence, the results of the study could only be appropriate for the situation that liver cancer cases were originated from cirrhosis. The data of GSE64041 were uploaded by Makowska et al. [15]. Their study focused on the molecular classification of liver cancer and the subclass-specific gene expression patterns. The hub genes were not screened. However, in the present study, we screened the hub genes as biomarkers for liver cancer by mining the same data. The study goals between Makowska et al. [15] and the present study were different.
The genesis and progression of liver carcinoma is a complex process, in which multiple genes and pathways may play important roles. Using bioinformatics methods, we analyzed the microarray-based high throughput data. As a result, a number of DEGs were screened out. GO analysis showed that these DEGs were mainly enriched in items regarding cell cycle and metabolism. In the KEGG pathway analysis, the DEGs were mostly enriched in six pathways that happened to be related to cell cycle and cell metabolisms. The results of the pathway analysis were in accordance with those of the GO analysis, indicating that aberrant functions of cell cycle and cell metabolism might play a key role during the genesis and development of liver carcinoma.
To screen any hub genes that might play critical roles among the DEGs, we constructed the PPI network. Consequently, the network was established by using STRING and Cytoscape tools. The hub genes were listed in the order of the degree that illustrates the relations of any certain gene with other genes. The top 10 node proteins, including TOP2A, PRDM10, CDK1, AURKA, BUB1, PLK1, CDKN3, NCAPG, BUB1B and CCNA2, were selected. Using Oncomine evaluation, the mRNA expression of the ten genes was higher in liver carcinoma than that in normal liver tissues, respectively. Then, the data of the HPA database showed that the protein expressions of these genes were in line with their mRNA expressions. This result indicates that the top 10 hub genes/proteins were over-expressed in liver cancer, and thus might play a critical role in its genesis and progression.
TOP2A (topoisomerase II
To evaluate the prognostic values of the hub genes, a TCGA cohort was used. Among these ten hub genes, only two genes (PLK1 and CCNA2) showed their prognostic value. PLK1 (Polo-like kinase 1) has been reported to be an oncogene in liver cancer [40]. It is a cell cycle protein that plays various roles in promoting cell cycle progression, particularly regulating the mitotic spindle formation checkpoint at the M-phase [41]. Besides, it is a proviral host-factor for hepatitis B virus replication [42]. Thus, PLK1 has been regarded as a target for liver cancer research [43]. CCNA2 (Cyclin A2) is expressed at elevated levels from S phase until early mitosis [44], and might promote liver cancer formation [45]. Hence, suppression of CCNA2 by miR-22 shows the therapeutic merit in liver cancer [46]. Nevertheless, their prognostic value in liver carcinoma has rarely been reported in the literature. In the present study, the data showed that the patients with low PLK1/CCNA2 expression level had a longer overall survival time relative to those of patients with high PLK1/CCNA2 expression level, respectively, suggesting that PLK1 and CCNA2 may act prognostic factors for liver carcinoma. Moreover, they may also be used as potential therapeutic targets for liver cancer.
In conclusion, the present study provides preliminary research on the mechanisms underlying liver carcinoma genesis and development. Microarray-based data were retrieved and DEGs were screened out by bioinformatics methods. Then, the functions of the DEGs were annotated by GO analysis and pathway enrichment analysis. They were mainly enriched in cell cycle and cell metabolism-related pathways. Afterwards, ten hub genes out of the DEGs were screened out and analyzed. Both the mRNA and protein expression of the hub genes were over-expressed in liver carcinoma, compared with normal liver tissues. Among the hub genes, PLK1 and CCNA2 have been indicated to be prognostic factors for liver carcinoma. Overall, the results of the present study gave a valuable indication for liver cancer research. However, future validation experiments are warranted to confirm the results.
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-171160.