
Abstract
BACKGROUND:
Circular RNAs (circRNAs) play an important role in pathogenesis and development of hepatocellular carcinoma (HCC). However, circRNA expression profiles in hepatitis B Virus (HBV)-related HCC remain to be studied.
METHODS:
Total 13 HBV-related HCC patients were enrolled for study. Three HCC and 3 paired adjacent non-tumorous (NT) tissues from 3 patients were performed for microarray. Ten pairs of HCC tissues were used to verify the identified up-regulated and down-regulated circRNAs obtained from the microarray data by quantitative real-time reverse transcription PCR (qRT-PCR). Total RNA was isolated and treated with Rnase R to remove linear RNA, then hybridized to the array to screen for circRNAs. Bioinformatics analyses including clustering, differential expression, annotation of circRNA/microRNA (miRNA) interactions, Go analysis and KEGG pathway analysis, were performed.
RESULTS:
Based on the microarray data, we found significantly up-regulation of 24 circRNAs and down-regulation of 23 circRNAs in the HCC samples compared to NT samples (fold change
CONCLUSION:
In this study, we comprehensively explored the expression of differentially expressed circRNAs in HBV-related HCC, and our results indicate that circRNA_101764 may play an important role in the development of HCC.
Introduction
The clinical information in 13 enrolled subjects
The clinical information in 13 enrolled subjects
#Serum HBV-DNA level was tested by quantified real-time polymerase chain reaction (qPCR, COBAS® AmpliPrep®/COBAS® TaqMan, Roche Diagnostics, Germany). The HBV DNA was undetectability less 20 IU/ml. *The patient was treated using nucleoside(s) analogues (NAs) before diagnosis of hepatocellular carcinoma. **IHC: Immunohistochemical staining for Hepatitis B surface antigen (HBsAg), glypican-3 (GPC-3) and hepatocyte (Hepa) protein were routinely performed.
Sequences of oligonucleotides used for RT-qPCR
Annotation of 3 circRNAs and miRNA response elements (MREs)
Circular RNAs (circRNAs), widely expressed in tissue and developmental-stage specific patterns that regulate gene expression in mammals. CircRNAs are a novel class of widespread and diverse endogenous RNAs characterized by the presence of a covalent bond linking the 3
Chronic hepatitis B virus (HBV) infection is a dominant risk factor in the pathogenesis of hepatocellular carcinoma (HCC) [7]. HBV carcinogenesis through integrating into the host genome, leading to the widespread instability [8]. Aberrant expression of genes, which can involve RNA, is a key node for the occurrence and development of HCC [9]. However, the hepatic expression of circRNAs in HCC tissues remains fully unknown. Therefore, the aim of this study was to screen hepatic circRNA profiles to identify and verify aberrantly expressed circRNAs in HBV-related HCC tissues.
Patients and liver tissue samples
Total 13 HBV-related HCC patients were enrolled for study. The diagnosis of HCC was performed following AASLD guideline and confirmed by histopathology [10]. All the tissue samples were reviewed by two experienced pathologists. All patients were serum HBSAg positive except other etiologies. Three HCC and 3 paired adjacent non-tumorous (NT) tissues from 3 patients were performed for microarray. Ten of 13 HCC tissues were used to verify the identified up-regulated and down-regulated circRNAs obtained from the microarray data by quantitative real-time reverse transcription PCR (qRT-PCR). The specimens were surgical resections, then immediately preserved in liquid nitrogen. The NT tissues were defined as being 5 cm away from the edge of the tumor and contained no tumor cells when evaluated by an experienced pathologist. The protocol was approved by the Ethics Committee of Beijing Youan Hospital affiliated to Capital Medical University and was conducted in compliance with the Declaration of Helsinki. The written informed consent was obtained from each patient. Patient demographics and clinicopathological data are summarized in Table 1.
Total RNA extraction
Total RNA was extracted from the all the HCC tissues and paired adjacent NT tissues using TRIzol reagent (Invitrogen, Carlsbad, SC, USA), following the manufacturer’s instructions. Total RNA from each specimen was quantified using a NanoDrop ND-1000 spectrophotometer (Wilmington, DE, USA). RNA integrity and genomic DNA contamination were assessed using standard denaturing agarose gel electrophoresis, and the purity was estimated by the ratio of absorbance at 260 to 280 nm.
Different expression profiles of circRNA between HCC and NT tissues. The hierarchical clustering shows a distinguishable circRNA expression profiles among 3 HCC tissue and paired adjacent nontumorous tissue (A). Volcano Plots visualized the relationship between fold-changeand statistical significance (B). The vertical lines correspond to 2.0-fold up and down, and the horizontal line represents a 
Microarray and qRT-PCR analysis of selected circRNAs. The top 6 highest change circRNAs (3 up-regulate and 3 down-regulate) were chosen and presented (A). Selected circRNAs shows a distinguishable expression profiles among HCC tissue and NTs through hierarchical clustering (B). qRT-PCR analysis of selected up-regulate (C) and down-regulate (D) circRNAs. *
Total RNA was treated with Rnase R (Epicentre, Madison, WI, USA) to remove linear RNAs. Each sample was amplified and transcribed into fluorescent complementary RNA (cRNA) utilizing Arraystar Super RNA Labeling Kit. The labeled circRNAs were hybridized onto the Arraystar Human circRNA Array. The labeled circRNAs were purified using the RNeasy Mini Kit (Qiagen, Germantown, MD, USA). The labeled cRNAs (pmol Cy3/
Raw data collection and expression profiles of circRNAs
Scanned images were imported into GenePix Pro 6.0 software for grid alignment and raw data extraction. Quintile normalization and subsequent data processing were performed using the R software package. Then, low intensity filtering was performed, and circRNAs that were flagged as “expressed” (greater than 2 times the background standard deviation) in at least two out of six samples were retained for further analyses. Differentially expressed circRNAs with statistical significance between two groups were identified using a volcano plot and hierarchical clustering. The statistical significance of the difference be conveniently estimated by t test. CircRNAs having fold changes
Measurement of circRNAs by qRT-PCR
Based on the circRNA expression profiles, we choose six of the most obvious changes circRNAs for further verification, which included 3 up-regulated(circRNA_102814, 100381, and 103489) and 3 down-regulated (circRNA_101764, 100327, and 103361). Total RNA was extracted from the 10 pair validation HCC tissues using Trizol Reagent (Invitrogen, Carlsbad, SC, USA) and reversely transcribed into cDNA using Super Script TM III Reverse Tran-scriptase (Invitrogen, Carlsbad, SC, USA). The mRNA content was normalized to the housekeeping gene
Annotation of circRNA/microRNA interactions
CircRNA (circRNA_100381, 103489 and 101764)/ miRNA interactions were predicted with Arraystar miRNA target prediction software based on TargetScan and Miranda [11, 12]. All the results were presented in Table 3. Differentially expressed circRNAs were annotated in detail with the circRNA/miRNA interaction information.
MicroRNA-target genes prediction and network
To further investigate the functional roles of microRNAs, which predicted to interact with choose circRNA, putative targets of miRNAs were predicted by three soft wares: TargetScan, Mirbase and Miranda softwares. Predict the final result to take three overlapping part of the data to predict the result. MicroRNA – mRNA interaction analysis was conducted by Cytoscape to elucidate correlations between microRNA and target mRNA. The size of each node represents the number of putative microRNA functionally connected to each mRNA.
MicroRNA target genes bioinformation analysis
GO analysis was performed to explore the functional roles of target genes in terms of biological processes (BP), cellular components (CC) and molecular functions (MF). Pathway analysis is a functional analysis mapping genes to Kyoto Encyclopedia of Genes and Genomes (KEGG). The P-value (EASE-score, Fisher-P value or Hypergeometric-P value) denotes the significance of the pathway correlated to the conditions.
GO analysis of predicted mRNAs according to the values in the enrichment score under the theme of BP, CC and MF.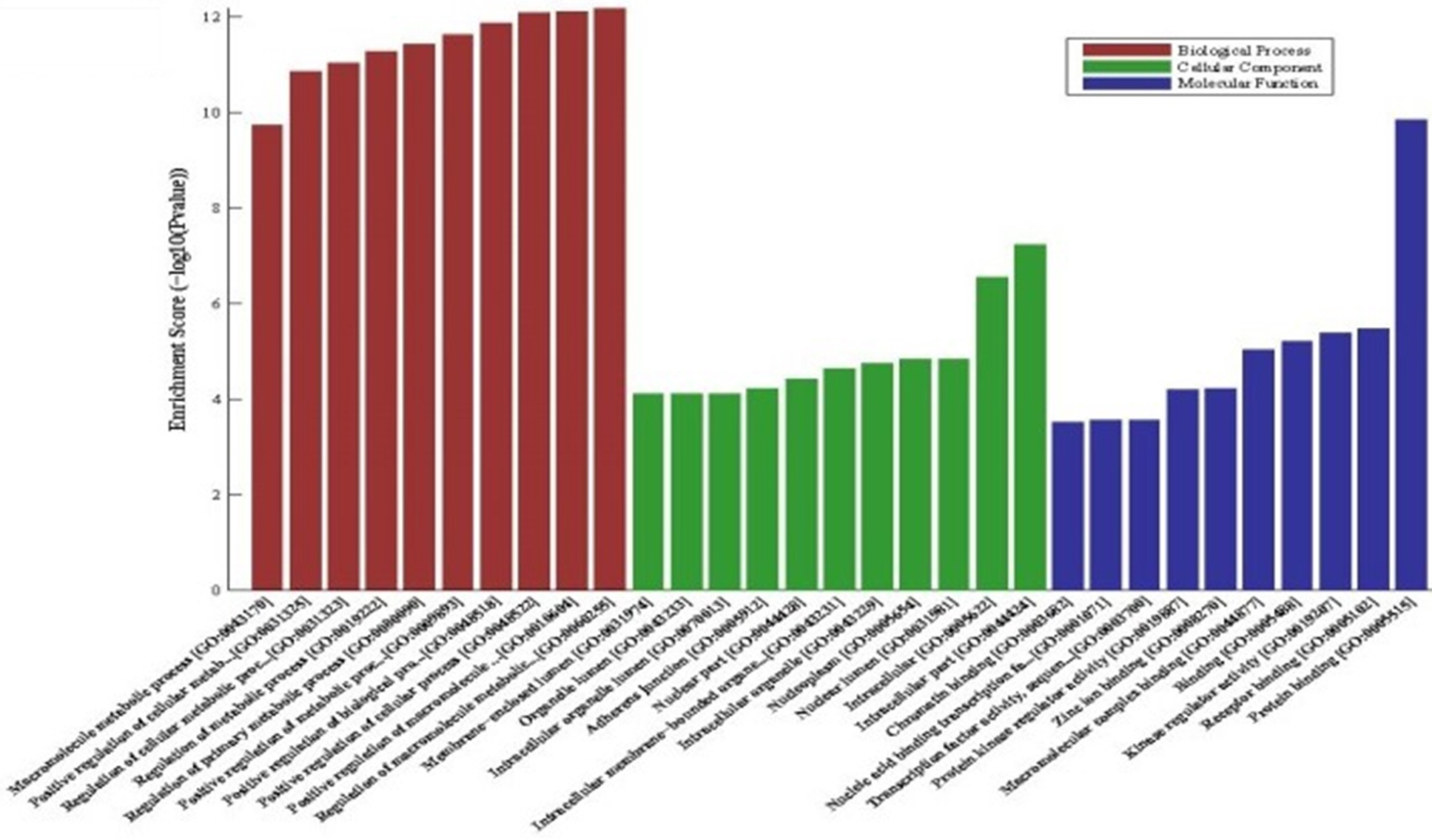
All statistical data were analyzed using SPSS v. 21.0 (SPSS, USA). All the results were reported as mean
Results
Different expression profiles of circRNA between HCC and NT tissues
A total of 5396 circRNAs were scanned under the microarray analysis, and the hierarchical clustering results show that there is a distinguishable circRNA expression profiles between HCC tissues and NTs (Fig. 1A). Volcano plots which are useful for visualizing differential expression between two different groups results exhibits that there were 24 up-regulated and 23 down-regulated cicrRNAs in all the tissues (Fold changes
Verification of selected circRNAs
Based on fold change, we choose the top 6 highest change circRNAs summarized in Fig. 2A. Hierarchical cluster analysis shows that chose circaRNAs were significant expression when comparing NTs (Fig. 2B). To further verify the microarray results, qRT-PCR was applied among the 10 HBV-related HCC tissues and paired NTs. The circRNA_102814 had the highest increased fold of 11.121 in microarray, but not significance in qRT-PCR(Fold changes
Pathway analysis of predicted mRNAs. The dot plot shows the gene ratio value of the top ten most significant enrichment pathways.
CircRNA/miRNA interactions and miRNA/mRNA network. The networke shown the relationship of miRNA181 family with target mRNA. Bule nodes are target mRNA, and red nodes are miRNA.
The significantly dys-regulated circRNAs were further categorized into different subgroups according to their genomic positions and effects. There were 1 antisense and 23 exonic circRNAs of increased circRNAs and 21 exonic and 2 intronic circRNAs of decreased circRNAs (data not shown). Verified three cicrRNAs then predict mRNAs to further analysis. The results of GO analysis of these mRNAs according to the values in the enrichment score under the theme of BP (Biological process), CC (cellular component) and MF(molecular function), where the top 3 processes were macromolecule metabolic process, macromolecule metabolic process and cellular process in BP; intracellular part, intracellular and nuclear lumen in CC; protein binding, receptor binding and kinase regulator activity in MF (Fig. 3).
Pathway analysis of predicted mRNAs
Thirty-four KEGG pathways were identified in all the mRNAs (data not shown). The top 10 KEGG pathways were shown in Fig. 4 based on GeneRatio. KEGG results shows that pathway in cancer was associated with 23 genes, which was associated with the largest number of genes.
Potential of circRNA/miRNA interactions and miRNA/mRNA network
All the three circRNAs (circRNA_101764, 100381 and 103489) were annotated in detail with circRNA/ miRNA interactions using Arraystar miRNA target prediction software (Table 3). It is interesting that circRNA_101764 mainly interact with miRNA181 family. The miRNA/mRNA network shows the relationship of miRNA with its target genes (data not shown). In consideration of miRNA181 family is very large, so we just show the miRNA181 family network in the Fig. 5.
Discussion
As we known that the most of the species in the transfer of genetic information are subject to the central dogma, which is DNA translated into protein via transcription into RNA. CircRNA, as is the case in general with non-coding RNAs, the question to answer is, “what do they do?” Individual examples of circRNAs have been sparsely described in the early 1990s. The examples of functional circRNAs were firstly found to act as microRNA sponges in 2013. CircRNA expression is a feature of essentially all eukaryotes known today [13]. Increasing experments suggests that circRNAs are involved in many disease, including cancer [14]. In currently, therefore, circRNAs emerging functions information on the mechanism of circRNA biogenesis, regulation and expression patterns are implicating them in multiple aspects of biology and disease, including potential roles in cancer, heart disease, synaptic transmission, and aging. Based on circRNAs was more stable than miRNA and LncRNA and therefore, they may be promising as diagnostic or predictive biomarkers [15].
The data gathered in this study suggest that prolfiles of circRNAs were very different between HCC and NT tissues. Based on the microarray data, we found significant up-regulation of 24 circRNAs and down-regulation of 23 circRNAs in the HCC samples compared to NT samples. Of them, hsa_circRNA 100381, 103489 upregulation and 101764 downregulation were firstly found to be significantly different. Fu et al. group found that circ_0004018 was significantly lower in HCC compare with para-tumorous tissue. Further data were found that this circRNA might involve in HCC carcinogenesis and metastasis through binding with miR-30-5e [16]. Other group found that circ_0001649 was significantly downregulated in HCC tissues compaire with para-tumorous tissue [17]. And Shang et al. group found that circ_0005075 could as a new potential HCC biomarker [18]. Huang et al. group found another cirRNA (circRNA_100338) was specific high expression in correlated with HBV-related HCC. The further data showed that CircRNA_100338 regulated of HCC invasion through sponging with miR-141-3p [19]. And in this study, we did not detect these circRNAs, maybe it’s because we used HCC tissues were all HBV-realted HCC and different circRNA chips.
CicrRNA research hot spot after one interesting found, which is the function of circRNAs mainly through be miRNA sponges to adsorption miRNA to affect the miRNA function [20]. Then some recent studies have demonstrated that circRNAs regulate gene expression by acting as competing endogenous RNAs, and verified they contain multiple, tandem miRNA binding sites [21]. In our study, bioinformatics analysis was used to annotate MREs and predict target mRNA related to the abnormally expressed circRNAs. We found that 34 KEGG pathways were identified in all the mRNAs which shows that pathway in cancer was associated with 23 genes, which was associated with the largest number of genes. This result shows that the three selected cicrRNAs have close relationship with cancer.
When we annotated in detail with circRNA/miRNA interactions by prediction software, we found that circRNA_101764 mainly interact with miRNA181 family which was correlates with many liver diseases. Chen found that miRNA181 family was very correlates with histological features of progression and harbors the potential to emerge as a diagnostic biomarkers for all types of liver cirrhosis [22]. Another research found that miRNA181a decreased the OPN (Osteopontin) expression to inhibit the liver cancer cells transfer and invasive ability [23].
In this research, we have identified significant up-regulation of 24 circRNAs and down-regulation of 23 circRNAs in the HCC samples compared to NT samples. The hsa_circRNA 100381, 103489 and101764 were confirmed to be significantly different. Those circRNAmay represent potential novel biomarkers for HCC early diagnosis and prognosis. In the future, we will expand the sample number for revalidations these circRNAs as potential biomarkers and functional studies.
Footnotes
Acknowledgments
This work was supported by the National ScienceFund (grant no. 81401970), Capital Science and Technology Development Fund (2014-1-2181), BeijingMunicipal Administration of Hospitals Clinical Medicine Development of Special Funding (ZYLX201610), and the Beijing Municipal Administration of Hospitals’ Ascent Plan (DFL20151602).