
Abstract
Background
We aimed to elucidate the pathogenesis of allergic rhinitis by analyzing aberrantly expressed circular RNAs and exploring their diagnostic value.
Methods
Blood samples were collected from allergic rhinitis patients and healthy controls, and circular RNA microarray was performed. The expression of differentially expressed circular RNAs was verified via real-time quantitative reverse transcription polymerase chain reaction, and these RNAs were selected for Gene Ontology and Kyoto Encyclopedia of Genes and Genomes analyses. Spearman’s correlation analysis was used to evaluate the relationship between hsa_circRNA_0008999, hsa_circRNA_0059181, disease symptoms, and total serum immunoglobulin E levels.
Results
Our microarray results revealed the presence of 200 differentially expressed circular RNAs. Two differentially expressed circular RNAs (hsa_circRNA_0008999 and hsa_circRNA_0059181) were selected and verified via real-time quantitative reverse transcription polymerase chain reaction. Gene Ontology analysis showed that hsa_circRNA_0008999 and hsa_circRNA_0059181 were closely related to the positive regulation of ligase activity, proteasome regulatory particle, nicotinamide adenine dinucleotide phosphate binding, and nuclear factor kappa-B–inducing kinase/nuclear factor kappa-B signaling. Kyoto Encyclopedia of Genes and Genomes analysis revealed the enrichment of proteasome, spinocerebellar ataxia, and nucleotide-binding oligomerization domain-like receptor signaling pathway. The circular RNA–microRNA–mRNA regulatory network suggested that hsa_circRNA_0008999 and hsa_circRNA_0059181 regulate the function and expression of microRNAs and mRNAs. The expression of hsa_circRNA_0059181 was correlated with the total symptom score and total serum immunoglobulin E levels in patients with allergic rhinitis.
Conclusion
We provided valuable information regarding the expression of hsa_circRNA_0059181, which may serve as a biomarker of allergic rhinitis.
Introduction
Allergic rhinitis (AR) is a consequence of allergic sensitization caused by immunoglobulin E (IgE)-mediated inflammatory reactions to environmental allergens and is characterized by nasal itching, sneezing, nasal obstruction, and nasal discharge. 1 In China, a cross-sectional population-based study showed that the prevalence of AR ranges from 8.7% to 24.1%. 2 Although AR is not a life-threatening disease, it imposes a heavy burden on the society and affects the academic performance of children. 3 Despite available treatment options, such as allergen avoidance, intranasal steroids, H1-antihistamines, leukotriene receptor antagonists, and allergen-specific immunotherapy, many patients are dissatisfied with the long-term effects of their treatment. In children, AR contributes to lost or insufficient time at school, decreased outdoor activities, sleep problems, otitis media, and rhinosinusitis. 1 As AR manifests with symptoms resembling those of a recurrent cold and is typically associated with the occurrence of asthma (50%–60% incidence), it remains commonly underdiagnosed.4,5 Hence, there is an urgent need to uncover the molecular mechanisms underlying the occurrence and progression of AR. 6
Circular RNAs (circRNAs) are a special type of endogenous noncoding RNAs (ncRNAs) that contain covalently closed-loop structures without a 5′-cap or 3′-poly-(A) tail. 7 At the molecular level, circRNAs can regulate the function of RNA-binding proteins, act as microRNA (miRNA) sponges, and modulate the expression level of their host genes. 8 Recent studies have shown that most circRNAs are conserved across species, stable and tolerant to exonucleases, and often exhibit tissue-/developmental-stage-specific expression. 9 Owing to these traits, the expression profile of circRNAs is considered a special molecular biomarker in various diseases such as cancer,10,11 cardiovascular diseases, 12 and allergic disease. 13 Zhu et al. 14 indicated that the overexpression of circular homeodomain-interacting protein kinase 3 increases the differentiation of T-helper 2 cells to CD4+ T cells, aggravating the nasal symptom scores in an AR mouse model. This finding suggested that circRNAs are involved in AR pathogenesis. Recent advances in high-throughput RNA sequencing and circRNA-specific computational tools have facilitated the identification and functional study of special circRNAs. 15
In the current study, we hypothesized that circRNAs might be involved in AR pathogenesis and could be crucial for the development of more effective diagnostic tools and therapeutic approaches. Thus, we explored the circRNA profiles of children with AR and revealed the important role of circRNAs in AR pathogenesis. The differentially expressed (DE) circRNA profile in the blood samples of children with AR was identified using circRNA microarray. Subsequently, the expression of the top two DE circRNAs was verified via real-time quantitative reverse transcription polymerase chain reaction (qRT–PCR) analysis. We further investigated the circRNA–miRNA–mRNA regulatory network using bioinformatics analysis. Finally, we identified the top two DE circRNAs as candidate serologic biomarkers for the diagnosis of AR.
Materials and methods
Patients and sample processing
The study was approved by the Medical Research Ethics Committee of Hangzhou First People’s Hospital (Hangzhou, China) (No. 2020-044-01, approval date: 24 December 2020). Patients diagnosed with AR who had undergone sinonasal surgery were enrolled. The standard criteria for AR diagnosis include major symptoms such as sneezing, clear runny nose, nasal congestion, and itching as well as serum specific IgE detection. All patients or their guardians provided a written informed consent before study enrollment. The present research was conducted according to the Helsinki Declaration of 1975, as revised in 2024. We obtained peripheral blood samples for circRNA chip screening from eight children, including four with AR (three boys and one girl; age range, 4–8 years) and four without AR (two boys and two girls; age range, 5–8 years) who visited the Hangzhou First People’s Hospital between January 2020 and March 2020. Peripheral blood samples (3 mL) were collected in ethylenediaminetetraacetic acid-containing anticoagulant tubes (Becton, Dickinson and Company, New Jersey, USA) and stored at −80°C until use.
Subsequently, 51 additional participants were recruited from the Department of Otorhinolaryngology of Hangzhou First People’s Hospital between January 2020 and December 2020, and peripheral blood samples were collected for further verification via real-time qRT–PCR analysis. All participants were children diagnosed with moderate-to-severe perennial AR, consistent with the 2008 Allergic Rhinitis and its Impact on Asthma guidelines. 16 All participants denied any history of allergic conditions or significant environmental exposures (such as chemical or air pollution).
The basic characteristics of all study participants are shown in Table 1. Nasal symptoms (nasal obstruction, sneezing, nasal discharge, nasal itching, and eye itching) were recorded using the visual analog scale (VAS), with scores ranging from 1 (slightest symptom) to 10 (worst symptom), and the sum of VAS scores was calculated as the total symptom score (TSS). All children stopped receiving any relevant medications for 1 month. Exclusion criteria included children with aspirin intolerance, asthma, and cystic fibrosis.
Characteristics of the study participants.

*TSSs (range: 1–50) were significantly different between the two groups (p < 0.001), as determined via one-way ANOVA.
AR: allergic rhinitis; TSS: total symptom score; ANOVA: analysis of variance.
RNA sample quality control
RNA extraction was performed using TRIzol (Invitrogen, Carlsbad, California, USA) according to the manufacturer’s instructions. The RNA concentrations of samples were determined via ultraviolet spectrophotometry using a NanoDrop ND-1000 instrument (Wilmington, DE, USA). RNA integrity was evaluated via standard electrophoresis on a denaturing agarose gel.
Labeling and hybridization
Labeling and array hybridization of the prepared samples were performed according to the manufacturer’s instructions (Arraystar Inc., Rockville, Maryland, USA). Briefly, total isolated RNA was initially treated with RNase R (Epicentre, Madison, WI, USA) to remove linear RNAs and enrich circRNAs. The collected circRNAs were then amplified and transcribed into fluorescent complementary RNA (cRNA) via a random priming method using a commercial kit (Arraystar Super RNA Labeling Kit; Arraystar) and purified using a RNeasy Mini Kit (Qiagen, Hiden, NRW, Germany). The specific activity and concentration of the labeled cRNAs (pmol Cy3/μg cRNA) were measured using a NanoDrop ND-1000 spectrophotometer. Then, 1 μg of each labeled cRNA was fragmented by adding 1 μL of 25× fragmentation buffer and 5 μL of 10× blocking agent, followed by heating at 60°C for 30 min. The samples were finally diluted with 25 μL of 2× hybridization buffer. The diluted solution (50 μL) was dispensed into the gasket slide and then assembled to the circRNA expression microarray slide. The chips were then incubated for 17 h at 65°C in an Agilent hybridization oven (Agilent Technologies, Palo Alto, CA, USA). After washing and fixing, the hybridized arrays were finally scanned with the G2505C Agilent microarray scanner.
Scanned images were imported using Agilent feature extraction software to extract raw data. These raw data were quantile normalized and processed using R software limma package (Vienna, Austria). After low-intensity filtering, circRNAs with flags in at least four of the eight present (P) or marginal (M) samples (“all targets value”) were chosen for further analyses.
DE circRNAs
The “fold change” between AR and control groups for each circRNA was calculated by comparing the differences in the profile of the two groups. Statistical differences in the profiles of the two groups were determined using the unpaired t-test. Accordingly, DE circRNAs were considered significant if fold changes were >2 and p-values were ≤0.05.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses
GO and KEGG pathway analyses were performed to predict the potential functions of significant DE circRNAs. The GO project provides a controlled vocabulary to elucidate the genetic regulatory networks of genes and gene products (http://www.geneontology.org). In particular, GO analysis comprises the following three domains: cellular component, molecular function, and biological process. KEGG analysis was performed to explore the significant signaling pathways associated with the selected DE circRNAs.
Construction of the circRNA–mRNA–miRNA regulatory network
To identify potential interactions between miRNAs and circRNAs, a home-made miRNA target prediction software based on TargetScan (http://www.targetscan.org/vert_72/) (“Weighted context++ score” <−0.1) and miRanda (http://www.microrna.org/microrna/) (“score” > 140, “Energy” <−1) databases was used to predict target miRNAs. The circRNA–mRNA–miRNA regulatory network was established and visualized using Cytoscape and custom python. The different colors and shapes of the nodes represented different RNA types and regulated relationships, respectively.
Real-time qRT–PCR
Total mRNA was extracted from blood samples using a commercial RNA extraction kit (TaKaRa, Osaka, Japan). The extracted RNA was converted to cDNA utilizing a reverse transcription kit (RR036A; TaKaRa) according to the manufacturer’s instructions. qPCR was performed using SYBR Green (RR420A; TaKaRa) as per the manufacturer’s instructions. All qRT–PCR assays were conducted using a Stepone plus Real-Time PCR system (Applied Biosystems, Waltham, MA, USA). Data were analyzed using the comparative threshold cycle (ΔΔCq) method.
17
All samples were tested in triplicate. The primers used were as follows:
β-actin (H), F: 5′-GTGGCCGAGGACTTTGATTG-3′, R: 5′-CCTGTAACAACGCATCTCATATT-3′; hsa_circ_0008999, F: 5′-AACCAACCATTGTTTGGAGTT-3′, R: 5′-TATACTGACAGCGTCATCTCTGT-3′; hsa_circ_0059181, F: 5′-CTTAGCTCCCTGTTCTTGGG-3′, R: 5′-GAGGAATCAAATTTCTTTCCAT-3′.
The thermocycling conditions used were as follows:
Step 1, initial denaturation: 95°C for 30 s; Step 2, amplification (40 cycles): 95°C for 5 s, 60°C for 30 s; Step 3, melt curve: 95°C for 10 s, 60°C for 60 s, 95°C for 15 s.
Total serum IgE (tIgE) measurement
Serum samples were collected and stored at −20°C until assay. The levels of tIgE were calculated using an ImmunoCAP system (Pharmacia Diagnostics AB, Uppsala, Sweden) according to the manufacturer’s instructions.
Statistical analysis
All statistical analyses were performed using SPSS version 16.0 (IBM Corp, Armonk, NY, USA), and the data were presented as mean ± standard error. Student’s t-test or one-way analysis of variance (ANOVA) were used to assess differences between the two groups. Spearman’s correlation analysis was used to assess the relationship between disease symptoms, tIgE, and chosen circRNAs. Differences were considered statistically significant when the p-value was <0.05 and false discovery rate was <0.05.
Results
Baseline characteristics of the study participants
The baseline characteristics of the study participants are presented in Table 1. The VAS scores of individual nasal symptoms ranged from 1 to 10, and TSSs were calculated as the sum of all nasal symptom VAS scores. The basic characteristics of the 59 children (8 of whom underwent circRNA chip screening) are shown in Table 1. The age of the participants ranged from 4 to 8 years, with a mean age of 6.58 years. We did not observe any statistically significant differences in age and sex between the two groups (p = 0.580 and p = 0.15, respectively). We revealed that the TSSs were 1.9 ± 1.73 in the control group and 15.14 ± 3.63 in the AR group (Table 1). Hence, the TSSs were significantly different between the two groups (p < 0.001).
Expression profiles of circRNAs in children with AR
We used a high-throughput circRNA microarray chip to investigate the expression profiles of circRNAs in children with AR. For this, we employed the Arraystar circular RNAs microarray chip (Arraystar Inc, Rockville, Maryland, USA), which contains thousands of microarray probes. Following labeling and hybridization, we analyzed eight blood samples using the microarray and identified the DE circRNAs with a fold change ≥2.0 and p-value <0.05. Then, we used the CircBase online database (https://www.circbase.org/) to delineate the details of circRNAs. Our results revealed that compared with the control group, the expression of 191 circRNAs was upregulated, while that of 9 circRNAs was downregulated in children with AR. The distributions of the expression profiles of circRNAs in all samples are shown in the box plot (Figure 1(a)). In addition, we used the scatter plot to assess the expression variations in circRNAs between the AR and control groups (Figure 1(b)). Moreover, we used the volcano plot to determine the statistical significance of alterations in circRNAs (Figure 1(c)). Finally, we performed hierarchical clustering to identify the relationship between the AR and control groups with regard to the expression patterns of circRNAs (Figure 1(d)).

circRNA profiles of all samples. (a) Box plot of circRNA expression. The abundance of circRNAs (ranging from maximum, upper quartile, median, lower quartile to minimum) is shown from top to bottom. (b) Scatter plot of circRNA expression. Variations in circRNA expression were assessed between the AR and control groups. (c) Volcano plot of the differentially expressed circRNAs between the control and AR groups. red, increased or decreased expression (p < 0.05) and (d) hierarchical clustering was performed to show the distinguishable expression patterns of circRNAs between the two groups. circRNA: circular RNA; AR: allergic rhinitis; Con: control group.
Real-time qRT–PCR validation of significantly DE circRNAs
Among the identified DE circRNAs in the microarray experiments, we selected two DE circRNAs for further studies. These circRNAs included a downregulated circRNA (hsa_circRNA_0008999) and an upregulated circRNA (hsa_circRNA_0059181). We further validated our results using peripheral blood samples obtained from children with and without AR (Figure 2(a)). The levels of tIgE among the participants are illustrated in Figure 2(b). Our results revealed that the relative expression of hsa_circRNA_0008999 was remarkably downregulated in the AR group compared with that in the control group (p < 0.01), whereas the expression of hsa_circRNA_0059181 was significantly upregulated in the AR group (p = 0.02).
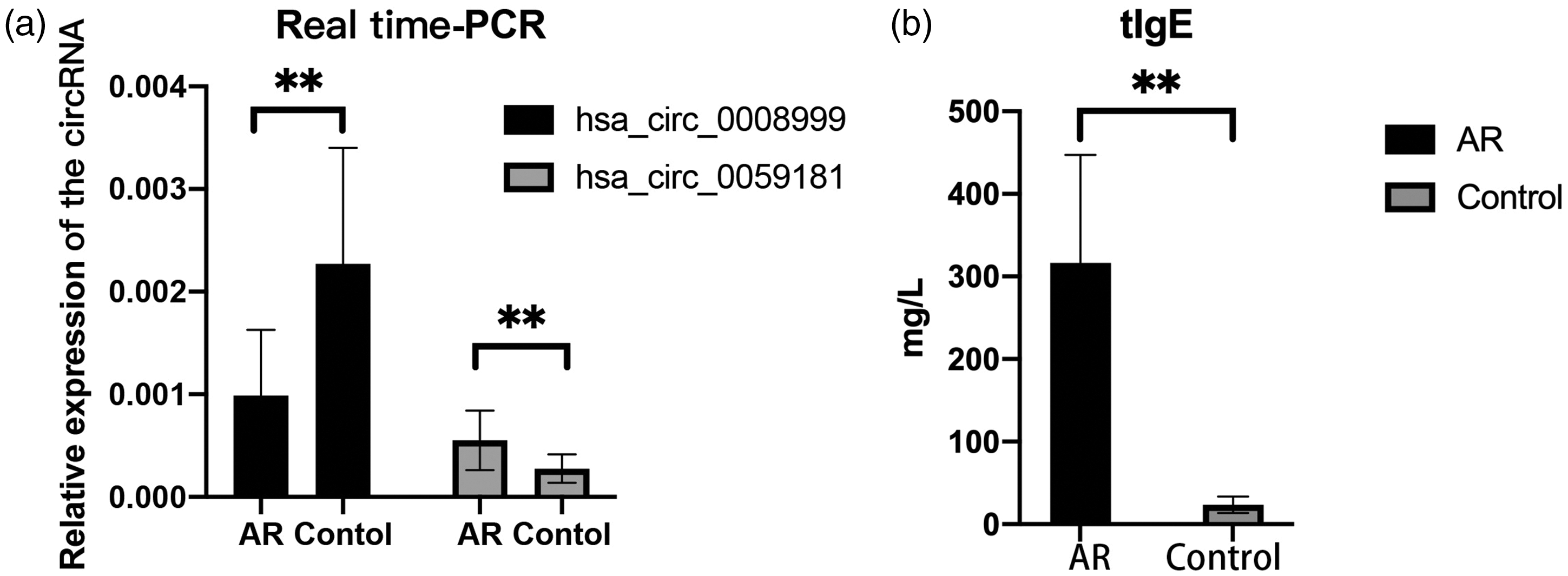
Validation of differentially expressed circRNAs and serum level of tIgE. (a) Real-time qRT–PCR. The expression levels of hsa_circRNA_0008999 and hsa_circRNA_0059181 were determined via circRNA sequencing. The relative expression level of hsa_circRNA_0008999 in the AR group decreased significantly compared with that in the control group (p < 0.01) and (b) the level of tIgE in the AR group increased significantly compared with that in the control group (p < 0.01). **p < 0.01 using unpaired t-test. AR: allergic rhinitis; circRNA: circular RNA; tIgE: total immunoglobulin E; qRT–PCR: quantitative reverse transcription polymerase chain reaction.
GO analysis of DE circRNAs
To investigate the molecular mechanism underlying the function of DE circRNAs that potentially participate in AR pathogenesis, we chose hsa_circRNA_0008999 and hsa_circRNA_0059181 and predicted their potential functions using GO enrichment and KEGG pathway analyses. GO analysis (Figure 3(a)) of DE genes revealed that all 123 genes were associated with the biological process domain. We revealed that the three most enriched biological process terms were “positive regulation of ligase activity,” “regulation of ligase activity,” and “snRNA metabolic process.” In addition, we identified a total of 31 genes that were associated with the cell composition domain. The three most enriched cell composition terms were “proteasome regulatory particle, base subcomplex,” “proteasome regulatory particle,” and “proteasome accessory complex.” Moreover, we revealed that 21 genes were associated with the molecular function domain. The three most enriched molecular function terms were “NADPH binding,” “acetylgalactosaminyl transferase activity,” and “NADP binding.”

Potential molecular function analyses via GO and KEGG pathway and the regulatory network of differentially expressed circRNAs. (a) GO functional classification map of the predicted genes regulated by differentially expressed circRNAs and (b) scatter plot of the enriched KEGG pathways. GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes.
The results of our KEGG pathway enrichment analysis are shown in Figure 3(b). We specifically noted the aberrant expression of genes in the “proteasome,” “spinocerebellar ataxia,” and “NOD-like receptor signaling pathway.”
Construction of circRNA–miRNA–mRNA regulatory network
We initially verified the expression of hsa_circRNA_0008999 and hsa_circRNA_0059181 using real-time qRT–PCR analysis and then selected them to construct a circRNA–miRNA–mRNA interaction network (Figure 4). Accordingly, we observed that the downregulated hsa_circRNA_0008999 was predicted to regulate the expression level of 16 miRNAs, while the upregulated hsa_circRNA_0059181 was predicted to regulate 14 miRNAs. To further explore the potential functions of these two DE circRNAs, we predicted the target mRNAs of these miRNAs (Figure 4). Our results suggested that circRNAs serve as miRNA sponges in AR patients.
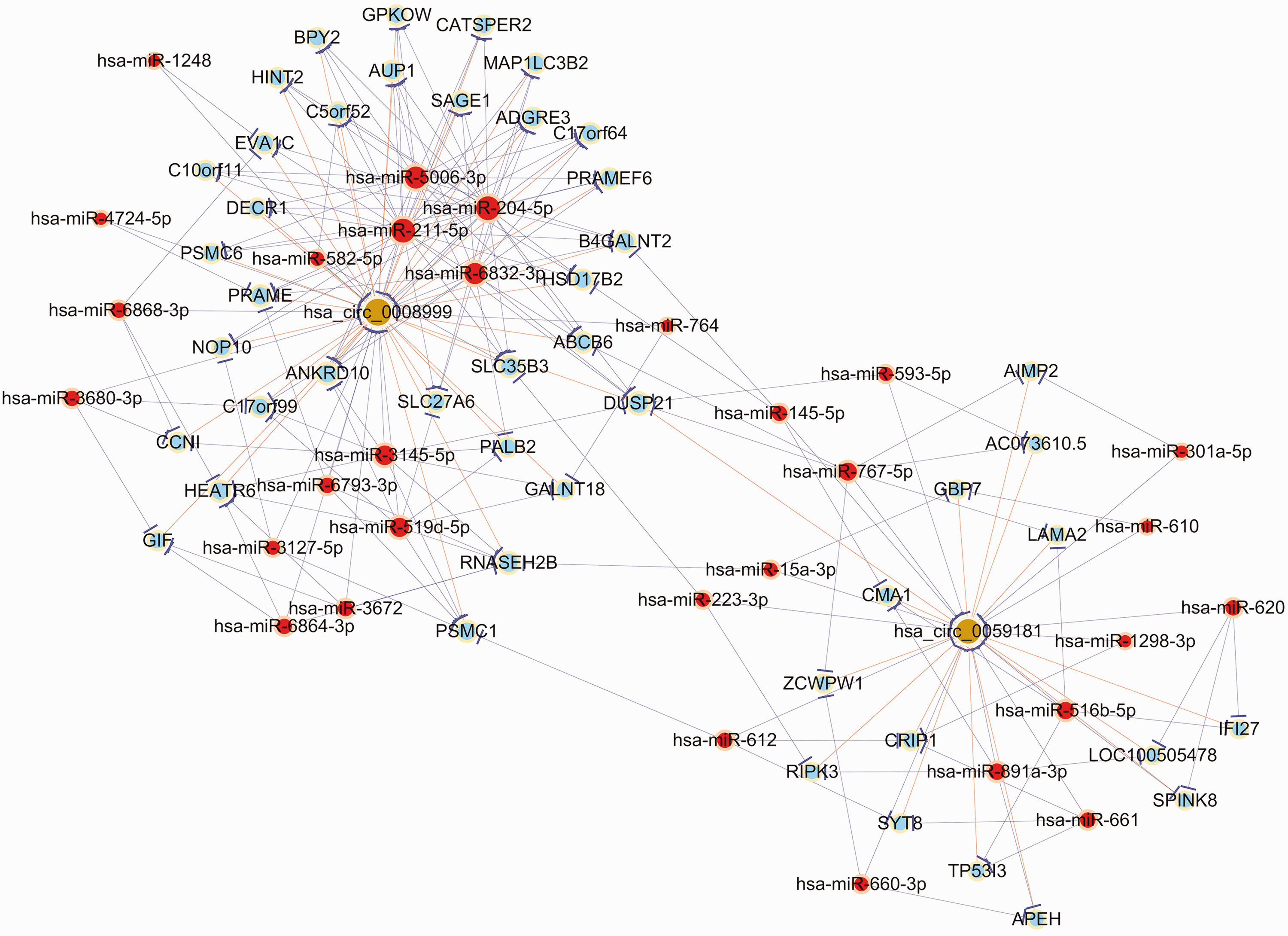
Prediction of the hsa_circRNA_0008999- and hsa_circRNA_0059181-targeted miRNA–mRNA network. Nodes with red color indicate microRNAs; those with light-blue color indicate protein-coding RNAs; and those with brown color indicate circular RNAs. Edges with a T-shape arrow represent directed relationships, and edges without an arrow represent undirected relationships. miRNA: microRNA.
Correlation of hsa_circRNA_0008999 and hsa_circRNA_0059181 with clinical scores and total IgE
We found that the expression levels of hsa_circRNA_0008999 and hsa_circRNA_0059181 were specific to children with AR. Therefore, we further analyzed the correlation between these circRNAs and the clinical characteristics of patients. We explored the feature of target circRNAs using the CircBase online database. We found that hsa_circRNA_0008999, which consists of 246 nucleotides, was located on exons 2 and 3 of the EED gene on chromosome 11, while hsa_circRNA_0059181 with a length of 278 nucleotides was located on exons 3 and 4 of the FKBP1A gene on chromosome 20. Moreover, we noticed that the low expression of hsa_circRNA_0008999 was neither correlated with the TSS (rho = 0.224, p = 0.242) nor with the level of tIgE (rho = 0.176, p = 0.361). However, we confirmed the correlation of the expression of hsa_circRNA_0059181 with TSS (rho = 0.673, p < 0.001) and tIgE (rho = 0.520, p = 0.004) in children with AR. Thus, hsa_circRNA_0059181 was identified as a potential biomarker in children with AR.
Discussion
Over the past several decades, the incidence of AR has been increasing worldwide, resulting in substantial economic and social burdens on patients and their families. 17 Accordingly, an increased trend has been observed in the prevalence of AR in children. 18 In particular, AR affects approximately 10%–40% of children worldwide. 5 Moreover, the coexistence of AR and asthma is common in children (with most studies reporting an incidence of 50%–60%). 5 The molecular mechanism of AR has not been fully elucidated. Accumulating research has indicated that circRNAs can act as miRNA sponges and regulate the expression of genes from the 3′-untranslated regions (3′-UTRs) of target mRNAs. Briefly, circRNAs are a novel class of ncRNAs with covalently closed-loop structures; circRNAs are highly conserved, stable, and abundant across species and specific diseases. 19 These special characteristics have drawn increasing attention to circRNAs, with an increasing number of researchers focusing on their diagnostic and therapeutic functions in many diseases, such as cancer, 20 systemic lupus erythematosus, 21 and respiratory and allergic diseases.22,23 For instance, Zhu et al. 14 showed that circARRDC3 could promote the differentiation of Th2 cells during AR development. In the current study, we aimed to explore the expression profiles of circRNAs in children with AR and identify the most significant circRNA biomarkers in the development of AR. Our results revealed the presence of 200 DE circRNAs (191 upregulated and 9 downregulated) (fold change ≥2.0, p < 0.05).
We specifically found that the regulatory network of circ_0059181 could target both miRNA (miR)-211-5p and miR-145. miR-211-5p has been recently reported to participate in the pathology of allergic asthma-induced pulmonary fibrosis. 24 Furthermore, miR-145 has been shown to regulate the balance of Th1/Th2 cells in allergic diseases. Interestingly, miR-145 has also been previously reported as a potential biomarker in the diagnosis and treatment of allergic asthma. 25 Therefore, these two miRNAs appear to be ideal circRNA candidates, and future studies should investigate their association with AR in children. Thus, we hypothesized that hsa_circRNA_0008999 and hsa_circRNA_0059181 might be involved in the development and pathogenesis of AR.
It has been reported that circRNAs can also act as miRNA sponges, regulating the expression of parental genes.10,19 Our GO term and KEGG pathway enrichment analyses (Figure 3) revealed the enrichment of nuclear factor kappa-B (NF-κB)–inducing kinase (NIK)/NF-κB signaling as well as the positive regulation of ligase activity and nucleotide-binding oligomerization domain (NOD)-like receptor signaling pathway in AR. NIK/NF-κB signaling is a multicellular transcription factor signaling pathway that plays a vital role in immune and inflammatory responses by regulating the expression levels of inflammatory-related cytokines and mediators in many diseases.26,27 NOD-like receptors (NLRs) are evolutionarily conserved families of intracellular pattern recognition receptors that play an important role in host physiology and innate immunity. 28 Genome-wide association studies of the NLR gene have identified numerous risk alleles in AR. 28 Therefore, we assumed that circRNAs might play an important role in the signaling pathways that participate in AR pathogenesis through the regulation of their gene expression.
Unlike linear RNAs, circular RNAs have a higher tolerance for exonuclease RNase R because of their circular covalently bonded structure. Owing to their abundance, conservation, and tissue specificity, circRNAs might act as specific molecular markers in many diseases.23,29
Our results showed that the expression levels of hsa_circRNA_0008999 and hsa_circRNA_0059181 were specific to children with AR. Despite this specificity, the expression of hsa_circRNA_0008999 was not specifically related to the clinical features (TSS) and tIgE of these patients. The host gene of hsa_circRNA_0008999 is EED. Studies have shown that the knockdown of EED alleviates the suppression of the positive glucocorticoid response element ((+) GRE), indirectly indicating that EED exerts anti-inflammatory effects. 30 Theoretically, hsa_circRNA_0008999 may modulate the inflammatory status in patients by regulating EED expression, potentially contributing to AR pathogenesis. Additionally, as previously mentioned, the target miRNA of hsa_circRNA_0008999 is associated with the NF-κB pathway. 31 Although our study did not identify a statistically significant correlation between hsa_circRNA_0008999 and clinical symptoms in patients, it remains a compelling candidate for further exploration in AR pathology.
However, the correlation between hsa_circRNA_0059181, TSS (rho = 0.673, p <0.001), and tIgE (rho = 0.520, P = 0.004) was specifically confirmed in children with AR. Thus, hsa_circRNA_0059181 was identified as a potential biomarker in children with AR. In the future, a multi-cohort validation should be conducted to confirm the value of hsa_circRNA_0008999. The development of diagnostic kits based on blood or nasal secretion samples, combined with hsa_circRNA_0008999 targeted biotherapeutics, may help elevate the clinical translational value of circRNAs. Further studies are required to explore the possible molecular mechanism of hsa_circ_0059181 in children with AR and verify whether hsa_circ_0059181 targets miRNAs and thus competitively sequesters the miRNA activity.
Currently, Nasal Allergen Provocation Test is the “gold standard” for the diagnosis of AR. However, this method carries the risk of acute allergic reactions, and the procedure is relatively complex. Other methods involve serum IgE quantification and skin prick tests, which are invasive. Furthermore, circRNAs hold substantial potential as diagnostic markers of AR in children. Moreover, circRNAs are known to exist in both blood plasma and exosomes owing to their high stability. 10 In addition, it is easy to obtain blood samples and perform an analysis, indicating that circRNAs have the potential to be a novel noninvasive biomarker in AR diagnosis. Future studies may reveal the increasingly important role of circRNAs in the diagnosis, prevention, and treatment of different diseases.
In conclusion, this study identified DE circRNAs between children with AR and healthy controls. Our findings suggested a positive association between the expression of circRNAs and AR. Future studies might employ functional assays to verify the role of DE circRNAs as miRNA sponges and their involvement in the development and pathogenesis of AR in mice as well as in humans. In this study, due to financial constraints, we could enroll only four study participants per cohort, which may have introduced bias in the results. Further large-scale investigations are required to validate these findings.
Footnotes
Acknowledgements
We would like to thank The Construction Fund of Key MedicalDisciplines of Hangzhou.
Author contributions
Jing Li and Yong Li conducted the studies, participated in collecting data, and drafted the manuscript. Yuhao Fan, Yilin Li, Miaomiao Xu, and Liya Ren performed statistical analysis and participated in its design. Yuhao Fan, Jing Li, and Yilin Li helped draft the manuscript. All authors have read and approved the final manuscript.
Consent to participate
All patients or their guardians provided written informed consent before enrollment to the study. The research was conducted according to the Helsinki Declaration of 1975, as revised in 2024.
Consent for publication
All patients or their guardians provided written informed consent for the publication of this study before enrollment.
Data availability
All data generated or analyzed during this study are included in this article. Further enquiries can be directed to the corresponding author.
Declaration of conflicting interest
The authors have no conflicts of interest to declare.
Ethical considerations
The study was approved by the Medical Research Ethics Committee of Hangzhou First People’s Hospital (Hangzhou, China) (No. 2020-044-01, approval date: 24 December 2020).
Funding
None.