
Abstract
Tyrosine kinase inhibitors (TKIs), a novel group of target-specific anti lung cancer drugs, have recently been found to resistant to some NSCLC cells which have the T790M EGFR mutation. However, recent investigations on the therapies of resistance to EGFR-TKIs are very limited. Therefore, it is important to develop more effective therapies to reverse EGFR-TKIs resistance. In our present study, erlotinib was used as the TKIs drug and the effects of the erlotinib on cell growth were evaluated. Cell viability and concentration dependent studies were performed using HCI-H1975 and HCI-H1299 cells alone with erlotinib, respectively. Further combined with rituximab, the results showed that erlotinib and rituximab were significantly inhibited the cell growth. Furthermore, the combination of erlotinib and rituximab greatly decreased the expression of p-mTOR and p-EGFR. Additional results from western blotting and immunofluorescence assays demonstrated that the accumulation of rictor was also decreased on MAM. Thus, all these results suggested that EGFR-TKIs combined with CD20 mono-antibody significantly decrease the cell growth of H1975 cells and H1299, with T790M EGFR mutation, and inhibit the localization of the key mTOR pathway proteins to MAM. So, it may be a promising strategy for overcoming EGFR TKI resistance in NSCLC patients.
Introduction
EGFR is a receptor tyrosine kinases (RTKs), facilitate tumorigenic abnormal signaling, which is highly expressed in NSCLC [1, 2]. Several tyrosine kinase inhibitor (TKI) therapies against EGFR is currently administered and is initially effective in L858R mutations NSCLC patients [3, 4, 5, 6, 7]. However, the overall efficacies of TKIs are limited due to the development of drug resistance [8, 9, 10]. T790M mutations may cause primary EGFR TKI resistance. However, attributed to the development of the T790M mutations, greater than 50% of NSCLC patients may acquire TKI resistance [2]. Therefore, it is important to elucidate the mechanisms of EGFR TKI resistance in order to reverse drug resistance and develop more effective therapies.
CD20 is an important differentiation antigen of B cells. It is firstly expressed in the pro/pre B cells. Most evidence indicates that CD20 is an important molecule that regulates the signal transduction and tumor drug resistance [11, 12, 13]. According to a series of biological response to produce CD20 and anti CD20 antibody binding, we indicated that CD20 may be involved in cell proliferation, differentiation, signal transduction and calcium ion transfer [14, 15, 16]. Since the intracellular region of CD20 is a non covalent binding with tyrosine protein kinase and serine/threonine protein kinase, CD20 and anti CD20 binding will stimulate the expression of these kinases such as EGFR [17]. However, the physiological role of CD20 has not yet been clarified.
Furthermore, mammalian target of rapamycin (mTOR) is a serine/threonine kinase, which plays a key role in the PI3K/Akt pathway [18]. mTOR kinase acts in two functionally different multi-protein complexes, mTOR complex 1 (mTORC1) and 2 (mTORC2), whose activities are regulated by complex co-factors. Recent discoveries suggest a distinct role for mTORC2 in cancer [9, 20]. Originally, mTORC1 regulates many cell growth factors to integrate mitogen to control cell proliferation. By contrast, mTORC2 has been shown to function as an important regulator of the cytoskeleton, which controls the phosphorylation of Akt through the interaction between rapamycin-insensitive companion of mTOR (rictor) [21, 22, 23]. Localization of mTORC2 in the cell has been suggested to be at the plasma membrane [24] But the function of mTORC2 at MAM is not clear.
This study demonstrates that combining rituximab with current EGFR-TKIs erlotinib may successfully inhibit cell proliferation in EGFR T790M-positive mutations NSCLC cells. Furthermore, in the case of T790M-positive cells, the localization of rictor is changed by combining rituximab with EGFR TKIs. This study suggests that the mechanism of rituximab to overcome resistance to EGFR inhibitor is altrering the location of rictor from the MAM. This indicates that selective combinatorial treatment should be used for cells with T790M-positive mutated EGFR, to improve NSCLC patient prognosis.
Materials and methods
Reagents and antibodies
Erlotinib [N-(3-ethynylphenyl)-6,7-bis(2-methoxy-ethoxy) quinazolin-4-amine] (Cat. No. E-4007) was purchased from LC Laboratories (Woburn, MA), rituximab [3,5,7,8-tetrahydro-2-[4-(trifluoromethyl)phenyl]-4H-thiopyrano [4,3-d]pyrimidin-4-one] (Cat. No. 53113) was purchased from Sigma-Aldrich (St. Louis, MO). The two inhibitors were suspended in DMSO and stored as aliquots at
Cell lines and cell culture
NCI-H1975 and NCI-H1299 cell lines were purchased from American Type Culture Collection (ATCC) (Rockville, MD, USA, CRL-5908 and CRL-5803, respectively). The NCI-H1752 and NCI-H1299 cells expressed both the L858R and T790M EGFR mutations (documented by ATCC). The two cell lines were all stored in incubators at 37
MTT cell viability assay
The effects of various TKIs on H1975 and H1299 cell viability were measured by MTT assay using MTT 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide dye according to the manufacturer’s instructions. Cells were seeded at 5000 cells per well on 96-well plates and after 24 hours, cells were treated with inhibitors for 72 hours. After added the MTT reagent (Sigma-Aldrich, MO, Cat. No. M5655), cells were incubated at 37
mTORC1 and mTORC C2 activities
Immunoprecipitations (IPs) of
Isolation of subcellular fractionation
Cells were washed twice in PBS and pelleted by centrifugation, resuspended and lysed using a motor-driven Potter-Elvehjem homogenizer. The supernatant from pelleted homogenates was then centrifuged at 10,000
Immunofluorescence
Thirty thousand cells were plated in cover glass slides in 12-well (Corning Glass Works, Corning, NY) per well. Cells were allowed to adhere for 24 hours. Cells were then treated with Erlotinib/rituximab. After fixing using 4% paraformaldehyde solution in PBS, cells were then permeabilized in 0.1% Triton X-100 solution and blocked in buffer containing 5% normal goat serum. Cells were incubated with Rictor primary antibody (1:400) at 4
Immunoblotting
Following the drug-treatments, cells were lysed in RIPA buffer (20 mM Tris, 150 mM NaCl, 10% sodium deoxycholate, 1% NP-40, 1 nM sodium orthovanadate, 10 mM sodium fluoride, and 1 mM phenylmethylsulfonyl fluoride, (Sigma-Aldrich). Cell lysates were then electrophoresed for separation on 10% SDS-PAGE. Separated proteins were then transfer to PVDF membrane for Western blot detection (Bio-Rad Laboatories, Hercules, CA) and incubated with specific EGDR or mTOR pathway related proteins antibodies overnight at 4
Statistical analyses
Data were expressed as mean
Rituximab induces sensitivity to erlotinib in NSCLC cells harboring T790M mutation EGFR. (A) IC
Comparison of IC
of NCI-H1975 cell lines and NCI-H1299 cell lines with NCI-H2170
It was confirmed that NCI-H1975 and NCI-H1299 cells were the T790M EGFR mutation cells. MTT cell viability assays were performed for determining the IC
IC50 of Erlotinib for NSCLC cell lines with and without T790M
IC50 of Erlotinib for NSCLC cell lines with and without T790M
Modulation of key mTOR proteins in TKI-resistant cells H1975 and H1299. (A) Western blot analysis expression of p-mTOR and p-EGFR after erlotinib or erlotinib combined with rituximab treatment; (B) After western blot analysis increased expression of EGFR and mTOR pathway related proteins in H2170-ER and H2170-SR cells were observed when compared to H2170-P cells with the exception of p-GSK-3Î, in H2170 SR cells. The fold changes were calculated using ImageJ software (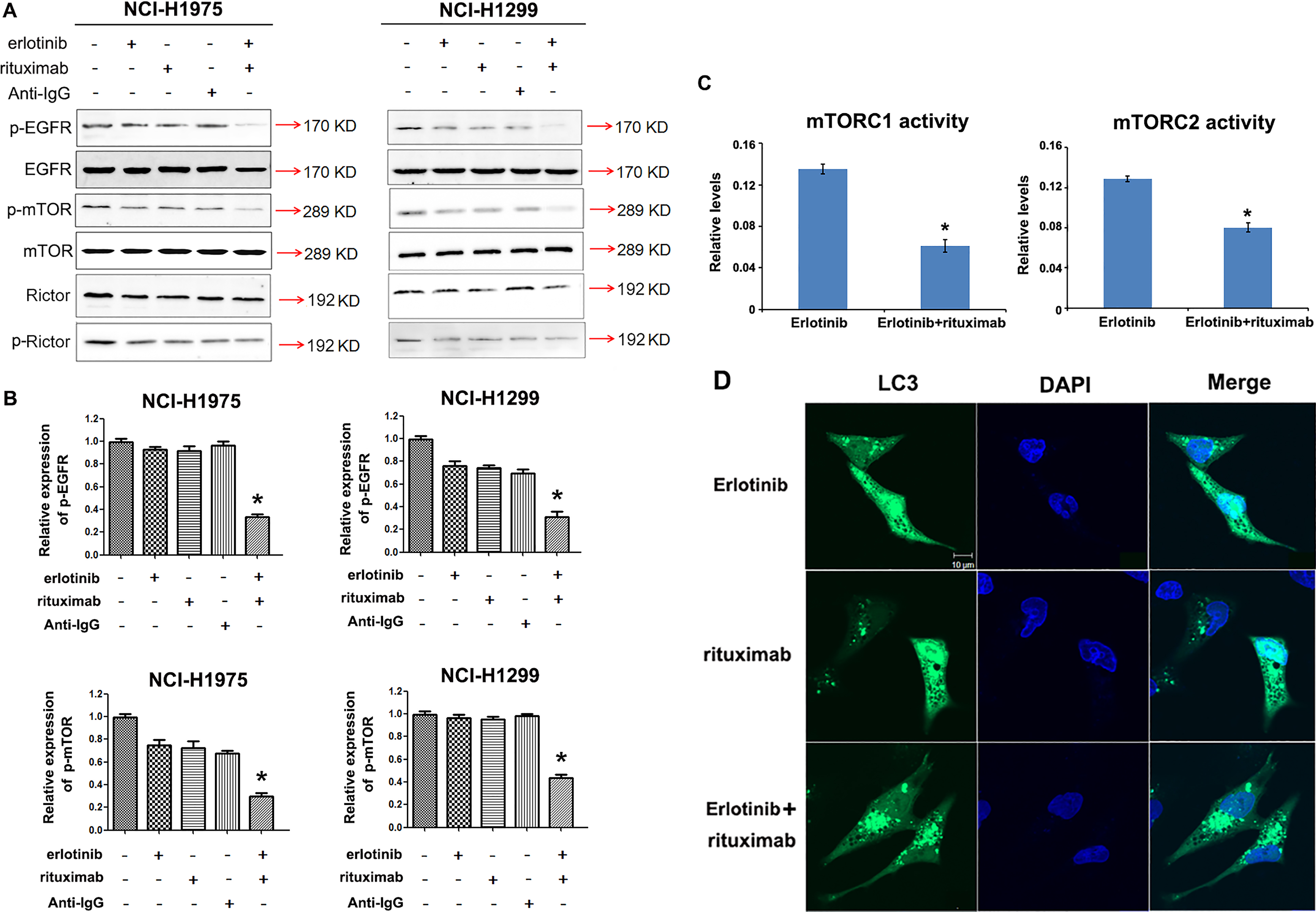
Since the T790M EGFR mutation is known to confer erlotinib resistance (Table 1) in NSCLC, it is important to find ways to reverse drug resistance caused by this mutation. It was suggested that CD20 may stimulate the exression of these kinases such as EGFR. Therefore, we tested for drug synergism on H1975 cells using rituximab, which is CD20 monoclonal antibody, via MTT cell viability assay. As shown in Fig. 1A, cell viability of both H1975 and H1299 cells were modestly inhibited by erlotinib and rituximab at lower concentrations but only erlotinib group. We confirmed the result by detecting the level of apoptosis marker PARP (Fig. 1B). Furthermore, we found that the synergistic effects on H1975 and H1299 cells growth inhibition were observed with erlotinib and rituximab in combination at concentrations of 2
Rituximab significantly restrains the localization of the component mTORC2. (A) mTORC2 component rictor is present in the ER fraction of cells. Total, whole cell lysate; cyto, cytoplasmic extract; ER, heavy membrane fraction from isopycnic flotation. Equal total protein levels were loaded in each lane. (B) Immunofluorescent staining with monoclonal anti- rictor antibody (green) in H1975 cells. ER/mito was stained by ER-RFP plasmid, F-actin was stained by using rhodamine-conjugated phalloidin (purplish red). DNA was counterstained by using DAPI (bule). Images were taken with laser scanning confocal microscope (Zeiss) with a 63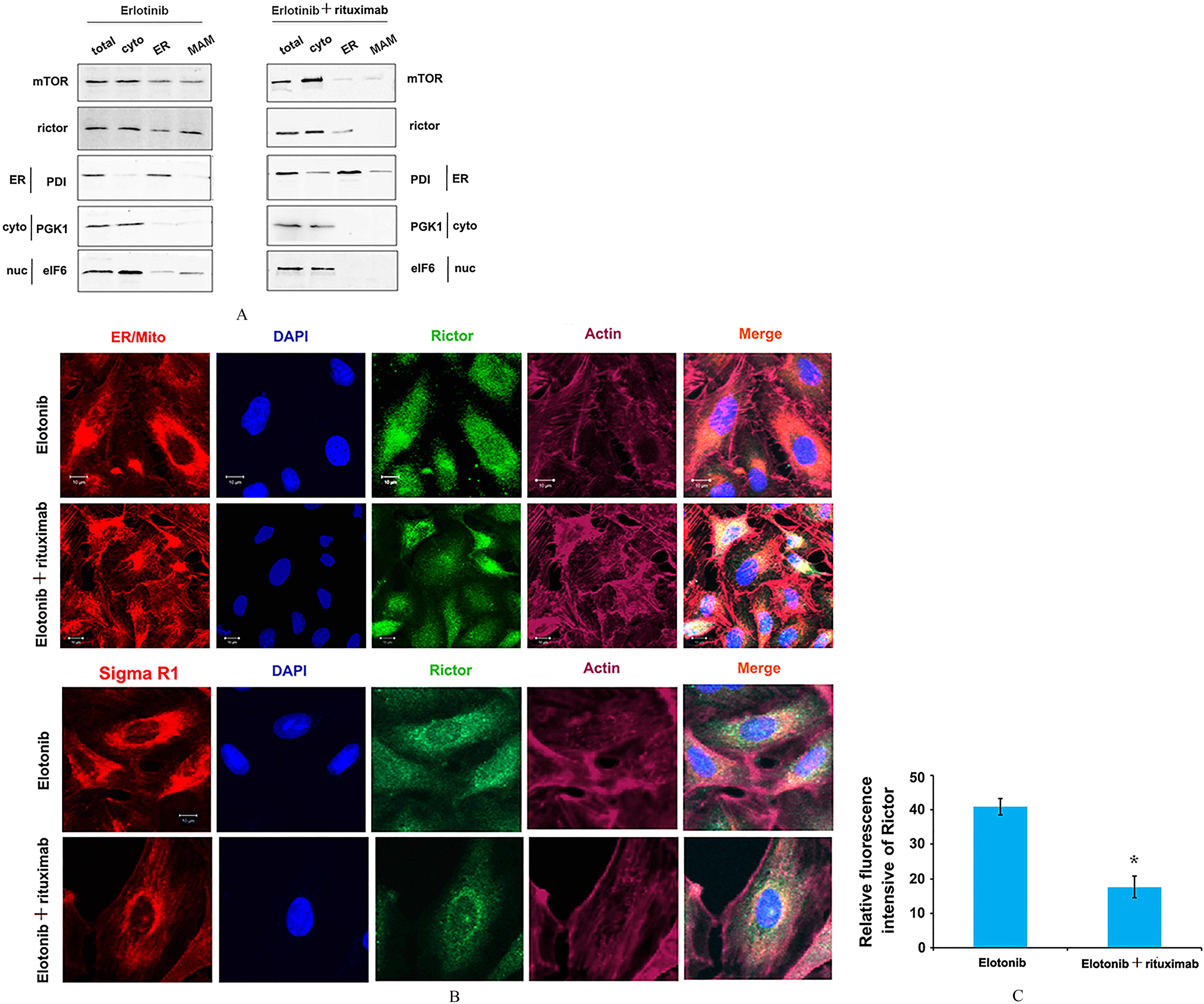
In order to elucidate mechanism of sensitivity to erlotinib combined with rituximab in TKI-resistance cells, expression levels of key proteins involved in mTOR/EGFR pathway were determined by immunob-lotting. We observed that active p-EGFR was downregulated 4-fold in the presence of erlotinib and rituximab respectively, in H1975 cells, when compared to only erlotinib group in H1975 cells. Furthermore, we observed that p-mTOR was also downregulated 3.0-fold in the presence erlotinib and rituximab in H1975 cells when compared to erlotinib treatment in H1975 cells. Similarly, in the H1299 cells, we observed p-EGFR was downregulated 4.0-fold in the presence erlotinib with rituximab; p-mTOR was downregulated 2.5 fold in the presence erlotinib with rituximab, when compared to a single agent of erlotinib treatments in H1299 cells (
To investigate whether the treatment with Erlotinib and rituximab induced the autophagy, we performed immunofluorescent staining sassay to detected the localization of LC3. We found that combined Erlotinib with rituximab could induce LC3 accumulation with the punctate structures characteristic of autophagy (Fig. 2D). These results indicated that Erlotinib with rituximab promote the recruitment and accumulation of LC3 and may be involved in the autophagy process.
Rituximab increased the sensitive to EGFR-TKI resistance cells by inhibiting mTORC2 localize to MAM
Previous studies have suggested that mTORC2 localize at the ER (39, 40), and the localization of mTORC2 to MAM is regulated by growth factors. So, we examined the localization of mTORC2 in the treatment of erlotinib with rituximab. First, we observed lower levels of mTORC2 (rictor) at MAM in H1975 cells upon erlotinib with rituximab stimulation compare with single erlotinib treatment (Fig. 3A). Second, we observed a significant decrease in colocalization of rictor with ER/mitochondria in H1975 and H1299 cells, grown in the presence of erlotinib and rituximab, compared with the cells upon erlotinib treatment only by immunofluorescence (Fig. 3B and 3C). These results suggested that the localization of mTORC2 to MAM is inhibited by the stimulation combined erlotinib and rituximab.
Discussion
To the best of our knowledge, the study was the first to discuss the combination treatment with EGFR-TKI and CD20 mono-antibodies to NSLCLs. However, studies have shown that EGFR phosphorylation was not inhibited by erlotinib in T790M EGFR mutation cells, accompanied by modestly promotion of cell proliferation [25, 26, 27]. So the lung cancer cells with T790M EGFR mutation were not highly sensitive to EGFR-TKIs. Indeed, the first generation of anti-EGFR therapies were all directed against the wild-type receptor. Based on the results of our study, lung cancers with T790M mutant EGFR are not sensitive to EGFR-TKI drugs, in contrast to wild-type EGFR cells. Thus, combining EGFR-TKI with the other drugs, which can reverse the resistance, might be a better therapeutic strategy for lung cancer. There is a voluminous literature that documents the successful use of CD20 monoclonal antibodies in the immunotherapy of cancer. Recent studies suggested that CD20 may stimulate the expression of these kinases such as EGFR [17]. However, the role of CD20 in the context of the EGFR-TKI setting remains unknown. In the present study, we determined the effect of the combination erlotinib with rituximab on the cell viability. The results suggest their combinatorial effects offer a promising strategy for EGFR-TKI resistance.
In our study, we measured the expression of the key proteins of mTOR during the drugs treatment. To identify the regulation of these proteins on EGFR-TKI resistance, we detected the expression of p-EGFR, p-mTOR and p-Rictor in EGFR-TKIs resistant cells H1975 and H1299 cells. There was a statistically significant down-regulation of p-EGFR and p-mTOR in the group which combine erlotinib with rituximab, compared to only erlotinib treatment (
In summary, we demonstrate that cell viability of lung cancer cells HCI-H1975 and NCI-H1299 with T790M EGFR mutation, treated with erlotinib and rituximab in combination at lower concentrations, which is not up to 10
Footnotes
Acknowledgments
This study was supported by Grands of Shanghai Council for Science and Technology (No. 15DZ19301 03).