
Abstract
Anaplastic lymphoma kinase (ALK) rearrangement is identified in 3–7% of advanced non-small cell lung cancer (NSCLC) cases, and ALK tyrosine kinase inhibitors (TKIs) have revolutionized the management of this subset of NSCLC patients. ALK–TKIs have been proven highly effective in ALK-rearranged advanced NSCLC patients, but after initial responses and benefit, a subsequent progression inevitably occurs. Understanding acquired-resistance mechanisms and defining an appropriate algorithm is becoming even more essential, particularly considering the availability of extremely efficacious next-generation ALK inhibitors. The aim of this review is the analysis of current data about ALK inhibition as a therapeutic strategy in ALK-rearranged NSCLC management, with a focus on a specific ALK–TKI, alectinib. Alectinib is a highly selective inhibitor of ALK and showed systemic and central nervous system (CNS) efficacy in the treatment of this particular population. The change of first-line approach, and consequently of further lines of therapy, in ALK-rearranged NSCLC patients is still a matter of debate. A summary of evidence from randomized trials evaluating alectinib will be presented in order to discuss the available clinical evidence, safety and place in therapy.
Keywords
Introduction
Lung cancer is the most frequently diagnosed tumour type and leading cause of cancer-related death, with 1.8 million new lung cancer cases estimated in 2012 and 1.59 million deaths worldwide, according to the recent statistics produced by the International Agency for Research on Cancer (IARC). 1
At the time of disease presentation, more than 50% of patients have evidence of advanced disease and, in these cases, median overall survival (OS) is less than 1 year, with 5-year relative survival remaining around 4%. 2 However, in the last few years, innovative drugs acting against therapeutic targets, initially for patients harbouring epidermal growth factor receptor (EGFR) mutations, then for those who present anaplastic lymphoma kinase (ALK) rearrangements, have dramatically improved prognosis of advanced non-small cell lung cancer (NSCLC) patients. 3
The aim of this review is to analyse current data about ALK inhibition as a therapeutic strategy in ALK-rearranged NSCLC management, with a focus on a specific ALK inhibitor, alectinib. A summary of evidence from randomized trials evaluating alectinib will be also presented in order to discuss the available clinical indications, safety and place in therapy.
ALK structure and diagnostics
The ALK gene is located on chromosome 2, and encodes for the ALK, a transmembrane receptor tyrosine kinase, and member of the insulin receptor super family. It was originally identified in 1994 in anaplastic large-cell lymphoma cell lines as a component of a chromosomal translocation, being normally expressed at low levels in adults, mainly in small intestine, testes and nervous system; this is probably due to a structural relation to its ligands, pleiotrophin and midkine, which play a role in the development of the nervous system and are known as the neurite growth-promoting family.4,5
ALK gene rearrangements were first identified in 2007 in 3–7% of NSCLC cases by two independent groups.6,7 This target is more commonly expressed in nonsmokers, younger patients, in acinus forms of adenocarcinomas of Asian patients and in signet-ring cell adenocarcinomas of White patients. It usually occurs independently of EGFR or KRAS (Kirsten rat sarcoma) mutations, and, in a few cases, it is not mutually exclusive, so patients with activating EGFR mutations should not be excluded from ALK screening. Finally, unlike EGFR mutations, it has a similar incidence across all regions of the world.8–10
The inversion in the short arm of chromosome 2 inv (2)(p21p23) results in the fusion of exons 1–13 of the echinoderm microtubule-associated protein-like 4 (EML4) gene with exons 20–29 of the ALK gene. However, the same ALK intracellular tyrosine kinase domain could fuse with different truncations of EML4-producing variants, all containing the entire intracellular kinase domain of ALK, encoded by exons 20 through to 29, but differing in the point of fusion with the EML4 gene. Among those, variant 1 (exon 13 of EML4 fuses with exon 20 of ALK in about 33% of cases), variant 2 (exon 20 of EML4 fuses with exon 20 of ALK in about 10% of cases) and variant 3a/b (exon 6 of EML4 fuses with exon 20 of ALK in about 29% of cases) account for more than 70% of lung cancers associated with EML4–ALK rearrangements.7,11,12 Moreover, in ALK-translocated NSCLC, EML4 does not appear to be the exclusive fusion partner with ALK, other less frequent fusion partners, such as TFG (TRK-fused gene), KIF5B (kinesin family member 5B) and KLC1 (kinesin light chain 1) have been described.13–15 EML4–ALK tyrosine kinases induce a ligand-independent dimerization of ALK, with a constitutive activation of downstream signalling pathways, including MAPK/ERK (mitogen activated kinase-like protein/extracellular signal-regulated kinases), PI3K/AKT (phosphatidylinositol-3-kinase/protein kinase B), and JAK/STAT (Janus kinase/signal transducer and activator of transcription), stimulating cell proliferation, differentiation and antiapoptosis. 4 Because ALK gene rearrangement involves a large chromosomal inversion and translocation, fluorescent in situ hybridization (FISH) assay using dual-labelled break-apart probes was initially the diagnostic gold standard approved by the US Food and Drug Administration (FDA).16–18 However, several reports also demonstrated a strong correlation between ALK immunohistochemistry (IHC) expression and ALK FISH test. For this reason, the VENTANA anti-ALK antibody (D5F3) was developed to maximize concordance with FISH in determination of ALK status, and as a consequence, FDA approved the VENTANA ALK (D5F3) CDx Assay (Ventana Medical Systems, Tucson, AZ) as companion diagnostics, recognizing IHC analysis as a diagnostic test for patient selection. Reverse-transcriptase polymerase chain reaction (RT-PCR) and next-generation sequencing (NGS) showed comparable performance with IHC when designed to detect the majority of fusions, and, according to Lindeman and colleagues’ indications, patients with positive results should be treated with an ALK inhibitor, although patients with negative results may benefit from a more sensitive method to exclude the possibility of a variant fusion.16,19 Similarly, amplicon-based NGS assays of DNA may fail to detect all fusion variants, thus a capture-based DNA or RNA approach is preferred for NGS testing for ALK fusions.16,20
Available therapeutic options in ALK-rearranged NSCLC patients and acquired resistance
For ALK-rearranged NSCLC patients, crizotinib (Xalkori®, Pfizer), a multitarget MET, ALK and ROS1-targeted tyrosine kinase inhibitor (TKI), received accelerated approval from the US FDA, and confirmed its efficacy in a frontline phase III trial (PROFILE 1014). Crizotinib 250 mg twice daily was compared directly with cisplatin or carboplatin plus pemetrexed showing a progression-free survival (PFS) benefit of 10.9 versus 7 months (hazard ratio, HR: 0.45; 95% confidence interval, CI: 0.35–0.60, p < 0.0001) and an objective response rate (ORR) equal to 74% versus 45% with chemotherapy.21–24
Although first-generation ALK inhibitor crizotinib is active with 57–74% ORR, most patients progress within the first year, with a median duration of response of 11.3 months, the central nervous system (CNS) being the most frequent site of progression. 25 Development of resistance to ALK–TKIs is currently a matter of evaluation and includes: (a) ALK-dependent mechanisms: where cell dependency on ALK signalling persists, even with ALK secondary resistance mutations or amplification; (b) ALK-independent ones: activation of bypass signalling pathways or drug efflux pumps such as P-glycoprotein (P-gp) which is a highly conserved adenosine triphosphate (ATP)-dependent efflux pump encoded by the multidrug-resistant 1 (MDR1) gene; and (c) phenotypic changes such as epithelial-to-mesenchymal transition (EMT) and small cell lung cancer (SCLC) transformation. 26
ALK resistance mutations appear to be one of the principal mechanisms of resistance and, unlike EGFR-mutant NSCLC where the T790M gatekeeper mutation is predominant, a much broader panel of on-target mutations has been identified in ALK-positive NSCLC treated with ALK–TKIs: for instance, substitution of glycine to arginine at codon 2032 in ROS1 kinase domain (G2032R) has been related to crizotinib-acquired resistance; G1202R ALK mutation causes resistance not only to crizotinib but also to next-generation ALK–TKIs tested in contrast to the L1196M mutation, the ‘gatekeeper mutation’ that hinders crizotinib binding at its active site on ALK, but remains sensitive to alectinib. 27 The amplification of wild-type EML4–ALK or ALK fusion gene amplifications (about 13%) lead to acquired drug resistance with or without concurrent ALK mutations (concomitant ALK–CNG and ALK–G1269A mutations were reported in one patient).17,28 In about 50% of ALK-rearranged NSCLCs, acquired resistance depends on activation of alternative downstream signalling pathways, including EGFR, HSP90 (heat-shock protein90), PI3K/AKT/mTOR (PI3K/AKT/mammalian target of rapamycin) or RAS/MEK (Rat sarcoma/Mitogen-Activated Protein Kinase) pathways, overexpression of phospho-ALK, phospho-EGFR, phospho-HER3 (human epidermal growth factor receptor 3), and phospho-IGFR-1R (insulin-like growth factor-1 receptor), KRAS mutations and KIT (KIT proto-oncogene receptor tyrosine kinase) amplifications.26,29 For these reasons, the development of next-generation ALK inhibitors against acquired resistance was encouraged, and alectinib (CH5424802), ceritinib (LDK378) and brigatinib (AP26113), have recently obtained FDA approval for treatment of crizotinib refractory, ALK-rearranged NSCLC, and a breakthrough-therapy designation was confirmed for lorlatinib (PF-06463922) in this setting. 30 Other ALK inhibitors, such as entrectinib (RXDX-101) and ensartinib (X-396), are currently under clinical development. 31
Alectinib: pharmacology
Alectinib (RO5424802/CH5424802) is a second-generation, ATP-competitive, orally and highly selective inhibitor of ALK, specifically designed to overcome crizotinib resistance. It presents a threefold increase in in vitro ALK inhibition compared with crizotinib (53 nmol/l versus 150.8 nmol/l). Unlike crizotinib, alectinib does not inhibit MET or ROS1 kinase activity, but it inhibits RET with comparable potency of ALK. Alectinib is effective, in vitro, in treating numerous crizotinib-resistant ALK mutations, including L1196M, F1174L, R1275Q, and C1156Y. It also showed in vitro efficacy against ceritinib-resistant ALK-mutant L1198F and moderate potency against the composite mutation D1203N+F1174C.32,33
Alectinib is metabolized by cytochrome P450 3A4 (CYP3A4), and its major active metabolite is M4. In patients with ALK-rearranged NSCLC treated twice daily with 600 mg oral alectinib, the geometric mean of maximum steady-state concentration (Cmax,ss) is 665 ng/ml for alectinib and 264 ng/ml for M4, suggesting a high exposure of the drug compared with crizotinib (steady-state concentration of 100–135 ng/ml). 33 Alectinib reached steady-state concentrations by day 7, with maximal concentrations at 4 h with an absolute bioavailability of 37% (90% CI, 34–40%) under fed conditions. 19 Overall, the pharmacokinetic profile of alectinib is comparable with its predecessors, crizotinib and ceritinib.34,35 The geometric mean elimination half-life is about 33 h. Alectinib and M4 do not inhibit CYP1A2, 2B6, 2C9, 2C19, or 2D6 and are predominantly excreted in faeces (97.8%). The coadministration of alectinib with a CYP3A inhibitor, a CYP3A inducer, or an acid-reducing agent (e.g. esomeprazole) seems not to generate clinically meaningful effects. 36
Alectinib, with its lipophilic properties, was intended to penetrate the CNS, since it effectively inhibits growth of ALK-positive CNS lesions in an intracranial tumour implantation model, reaching concentrations in the cerebral hemispheres and cerebellum tissue, comparable with those in the plasma. Unlike crizotinib and ceritinib, as evident in in vitro studies, alectinib is not a substrate of P-gp, the key efflux transporter that delays drug penetration through the blood–brain barrier (BBB). This could explain its higher ratio in cerebrospinal fluid (CSF) approaching 0.75 ng/ml (versus crizotinib CSF concentration of 0.616 ng/ml reported by Costa and colleagues 39 ) and corroborates the reported activity of alectinib also in ALK-positive NSCLC patients with leptomeningeal disease.37–39
Alectinib: clinical trials
Alectinib showed remarkable results in recent clinical trials and the most impressive of them are discussed subsequently and reported in Table 1.
Alectinib efficacy in selected and discussed clinical trials.
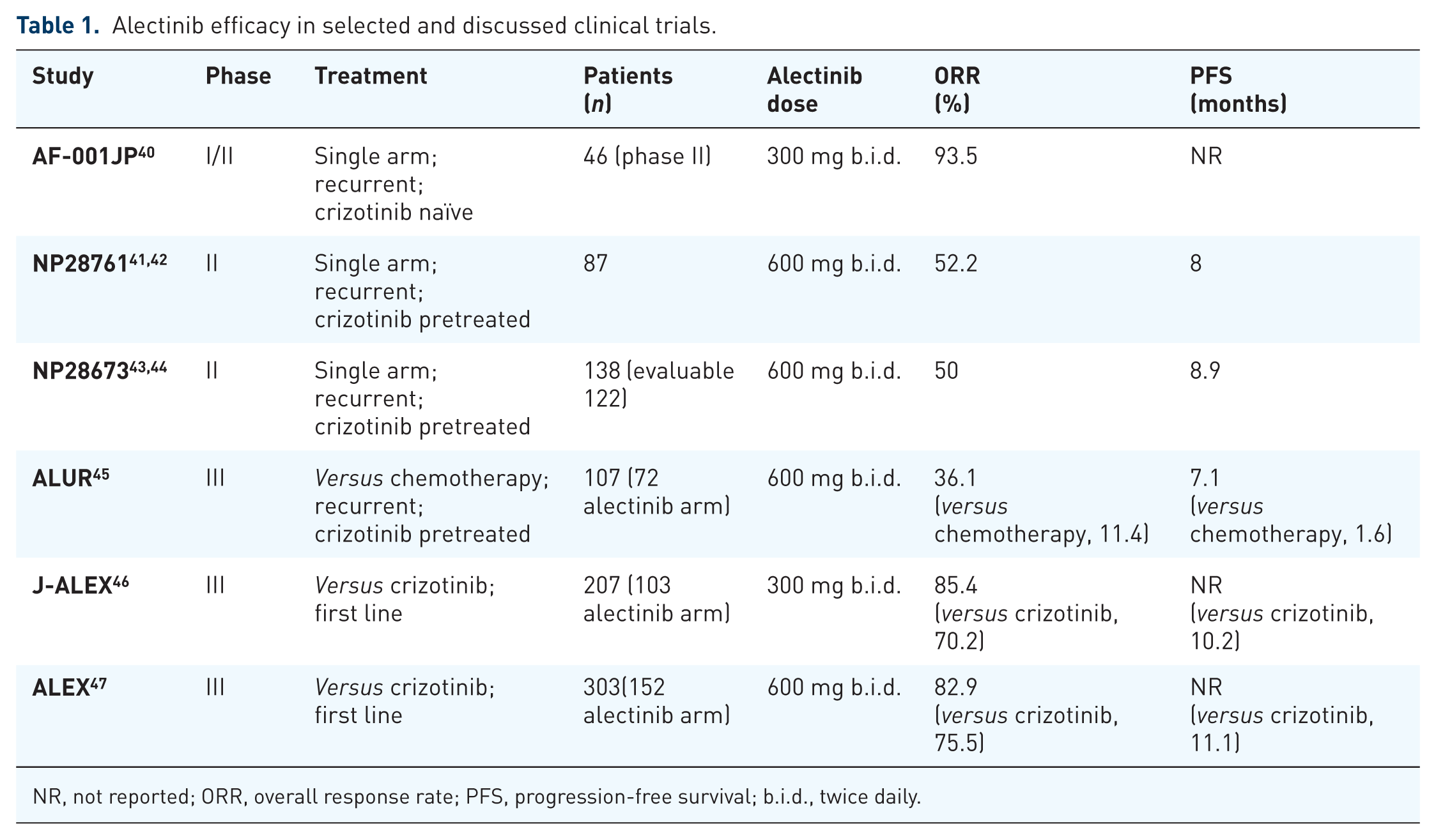
NR, not reported; ORR, overall response rate; PFS, progression-free survival; b.i.d., twice daily.
Alectinib was firstly evaluated in ALK-translocated crizotinib-naïve Japanese advanced NSCLC patients in a multicentre, single-arm, open-label, phase I/II study (AF-0001JP). Overall, 436 patients were screened for ALK; 135 (31%) were identified as ALK rearranged, but only 70 of them were then enrolled and treated in either phase I (24 patients) or phase II part (46 patients) of the study, as the remainder of them were considered ineligible by the investigators. In the phase I study, 24 patients received 20–300 mg oral doses twice daily. Dose-limiting toxicity (DLT), maximum tolerated dose (MTD) and pharmacokinetic parameters were primary endpoints, while in the phase II part, primary endpoint was objective response (OR). A total of 20 out of 24 patients presented measurable lesions showing an OR, according to investigators’ assessment, of 85%. No DLTs or grade 4 adverse events (AEs) were reported in the phase I trial, even with the highest dose, but further titration was precluded due to national limitations on exposure to excipients in the formulation. As a consequence, no MTD was identified, and 300 mg twice daily was the recommended dose for the phase II portion. Of 46 treated patients in the phase II part, OR was 93.5% (95% CI, 82.1–98.6), achieved in 43 of them.19,40 With 3 years’ follow up, 25 of 46 subjects were still under treatment, reaching a 3-year PFS rate of 62% (95% CI, 45–75) and a 3-year overall survival (OS) rate of 78%. 48
In the US, a phase I/II study (AF-002JG/NP28761) enrolled 47 ALK-positive NSCLC patients progressed on or intolerant to crizotinib. Doses of 300–900 mg twice daily were evaluated during the dose-escalation phase I part, aiming to find the recommended dose for the phase II portion. Dose escalation started at the highest-level dose defined by the Japanese study: no DLTs were observed up to the highest dose tested (900 mg twice daily) except for two patients who required dose modification due to grade 3 headache and neutropenia, respectively. On the contrary, alectinib, 600 mg twice daily, was associated with both clinical activity and good tolerability, becoming the recommended dose for phase II part. Of note, 44 patients were evaluable for alectinib activity with an investigator-confirmed ORR of 55%.19,37 The single-line multicentre phase II study (NP28761) was conducted in the US and Canada: 87 patients with ALK-positive NSCLC received alectinib 600 mg twice daily until progression, death, or withdrawal, after crizotinib progression. OR by an independent review committee (IRC) was the primary endpoint. 41 At the time of the updated analysis, a total of 67 patients had measurable disease at baseline, reporting an OR of 52.2% (95% CI, 39.7–64.6), with a median duration of response of 14.9 months. Median PFS in the entire population (n = 87) and OS were 8 and 22.7 months, respectively. 42
A phase II study (NP28673) similar to NP28761, showed activity of alectinib 600 mg orally twice daily in a cohort of 138 crizotinib-resistant, ALK-positive NSCLC patients with an OR of 50% (n = 122 patients evaluable for response by IRC), and a median PFS for all the entire population of 8.9 months (95% CI, 5.6–11.3 months).43,44
To date, at American Society of Clinical Oncology 2018, Ou and colleagues reported the final pooled phase II OS and safety data after a longer duration of follow up of the two single-arm, open-label phase II studies (North American NP28761 and global NP28673). 49 The pooled data set included 225 patients after a median follow up of 92.3 weeks. At the time of final data cut off, 53.3% of patients died, 39.1% of them were alive and in follow up, while 7.6% withdrew consent or were lost to follow up. Alectinib demonstrated a median OS of 29.1 months (95% CI, 21.3–39.0), specifically in NP28673 it was 29.2 months (95% CI, 21.5–44.4), while in NP28761 it was 27.9 months (95% CI, 17.2–not estimated, NE), respectively. 49
Previous data were further confirmed by the phase III ALUR study that investigated efficacy and safety of alectinib versus standard chemotherapy [pemetrexed 500 mg/m2 every 3 weeks (q3w) or docetaxel 75 mg/m2 q3w] at relapse on n = 107 ALK-positive NSCLC patients previously treated with platinum-based doublet chemotherapy and crizotinib. Alectinib presented systemic and CNS efficacy, including PFS and ORR, versus chemotherapy: median PFS by IRC was 7.1 months in the target therapy arm versus 1.6 months in chemotherapy one (HR 0.32; 95% CI, 0.17–0.59, p < 0.001) and ORR was 36.1% versus 11.4% (95% CI, 0.05–0.43), respectively. 45
Results from previous studies highlighted alectinib as an effective treatment strategy leading to planning its evaluation in phase III studies as first-line approach.
J-ALEX, a phase III study, compared efficacy and safety of alectinib versus crizotinib in Japanese ALK-positive advanced or recurrent NSCLC patients with no prior ALK inhibition therapy. In this trial, 207 patients were treated with either alectinib 300 mg twice daily, or crizotinib 250 mg twice daily. At the preplanned interim analysis, the study was stopped because the primary endpoint, PFS, was met (HR 0.34, p < 0.0001). Median PFS was not yet reached with alectinib (95% CI, 20.3 months–NE) and was 10.2 months with crizotinib (95% CI, 8.2–12). Furthermore, alectinib showed a higher proportion of patients achieving an OR (85.4% versus 70.2%) and a smaller proportion of grade 3 or 4 toxicity events (26% versus 52%) compared with crizotinib. 46
These impressive results were then confirmed by a second phase III trial, ALEX, comparing alectinib with crizotinib in ALK-translocated advanced NSCLC. A total of 303 patients were treated with either alectinib 600 mg twice daily or crizotinib 250 mg twice daily; of them, 55% were non-Asian. Stratification included brain metastasis, primary endpoint was PFS and secondary endpoint was time to CNS progression. Authors reported results similar to those already presented in J-ALEX, showing alectinib superiority compared with crizotinib: lower chance of progression (41% versus 68%), higher 12-month event-free survival rate (68.4% versus 48.7%), lower rate of CNS progression (12% versus 45%), higher response rate (82.9% versus 75.5%) and less AEs (41% versus 50%). Median PFS was not yet reached with alectinib (95% CI, 17.7 months–NE) and was 11.1 months with crizotinib (95% CI, 9.1–13.1). 47
Evaluating results particularly from the J-ALEX and ALEX trials, a specific consideration should be directed to dose selection, which has a critical role in maximizing the potential for efficacy, while minimizing the potential for safety risks in a target patient population. 50 Mok and colleagues, at European Society for Medical Oncology Asia 2017, presented data of Asian versus non-Asian pretreated ALK-positive patients from the global phase III ALEX study of alectinib (600 mg twice daily) versus crizotinib (250 mg twice daily) in first-line advanced ALK-positive NSCLC. Overall, 303 patients were randomized: alectinib n = 152 (n = 69 Asian, n = 83 non-Asian), crizotinib n = 151 (n = 69 Asian, n = 82 non-Asian). Efficacy and safety data were similar between subgroups, as well as AE profiles, confirming alectinib 600 mg twice daily dosage to be more effective than crizotinib in Asian and non-Asian patients, also having an acceptable safety profile in the first subgroup. 51
Alectinib: safety
According to published clinical trials, alectinib appeared to be safe with a good toxicity profile, as reported also in Table 2.
Alectinib safety in selected and discussed clinical trials.
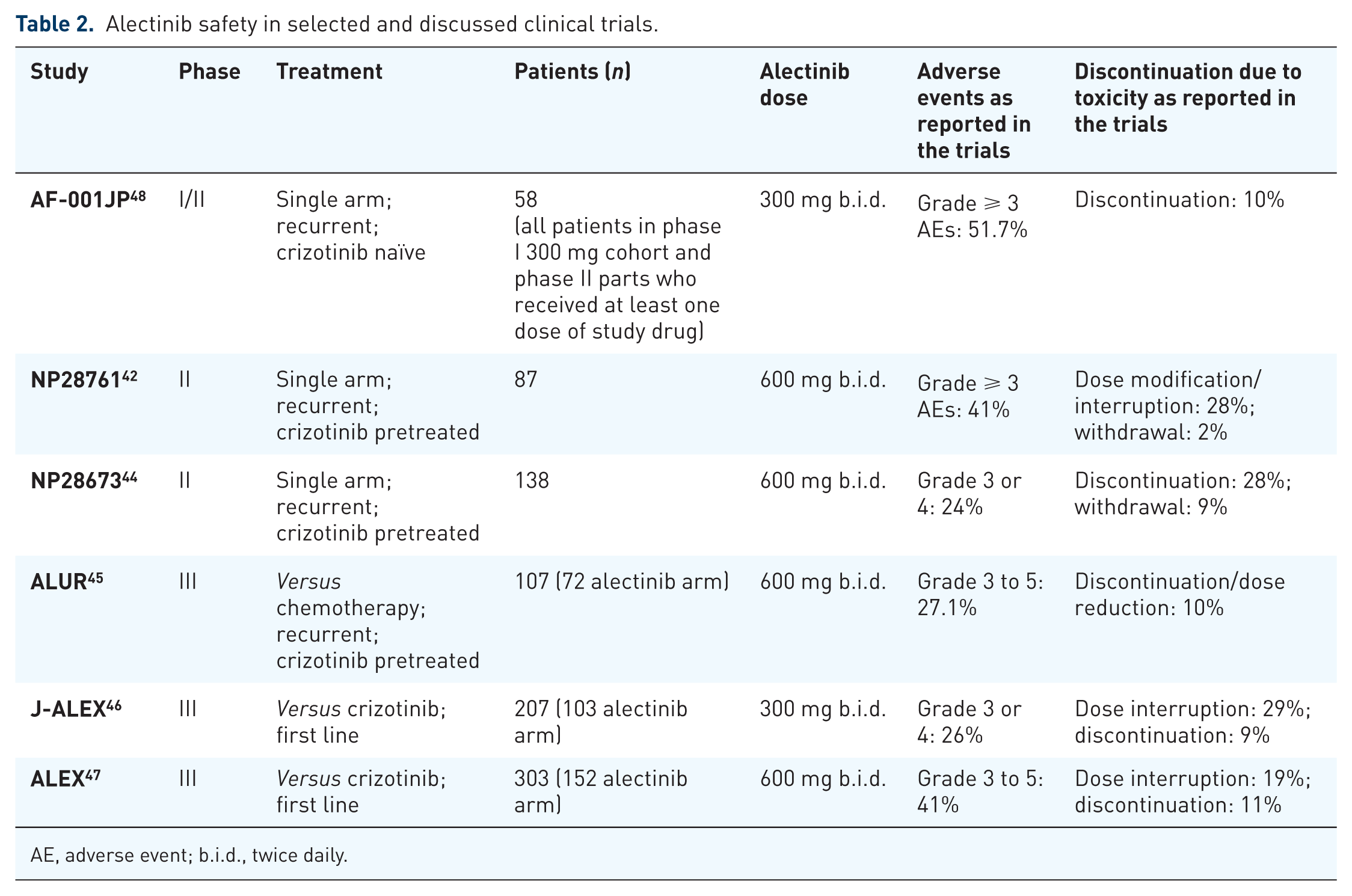
AE, adverse event; b.i.d., twice daily.
At last update, phase I/II Study AF-001JP reported treatment-related AEs in 56 patients (96.6%) on the safety population which comprised all patients in the phase I 300 mg cohort and phase II part, who received at least one dose of study drug (n = 58). Grade 3 AEs were reported in 16 patients (27.6%). There were no treatment-related grade 4 or 5 AEs, while the most common treatment-related AEs (all grades) were increased blood bilirubin (36.2%), dysgeusia (34.5%), increased aspartate aminotransferase (32.8%), increased blood creatinine (32.8%), and constipation (31.0%). Serious AEs were reported in 24.1%. 48
In the NP28761 study, the most common grade 1 or 2 AEs were constipation (36%), fatigue (33%), myalgia (24%) and peripheral oedema (23%); while the most common grade 3 or 4 AEs were creatine phosphokinase (CPK) elevation (8%), and alanine aminotransferase and aspartate aminotransferase elevation (6% and 5%, respectively). Two patients withdrew because of hepatotoxicity, 28% had AEs leading to dose modification/interruption. One patient died of haemorrhage that was considered to be related to treatment, however, this patient was also on concomitant anticoagulation. 42
In the NP28673 trial, constipation (38%), fatigue (31%) and peripheral oedema (30%) were the most common treatment-related AEs (any grade). Rates of dose interruption and withdrawal were 28% and 9%, respectively. 44
Updated pooled analysis of NP28761 and NP28673 demonstrated grade ⩾ 3 AEs (any cause) in 44% of patients treated with alectinib, while 14.7% and 37.3% of cases experienced AEs leading to dose reductions and dose interruptions/modifications, respectively, and, finally, 6.2% of them experienced AEs leading to withdrawal. 49
ALUR investigators discovered 77.1% AEs (all grades) in the alectinib arm and 85.3% in the chemotherapy one, with grade 3–5 AEs in 27.1% and 41.2%, respectively. One fatal AE was reported in the chemotherapy arm. AEs leading to discontinuation or dose reduction occurred in 10% and 20.6% of the alectinib and chemotherapy groups, respectively. 45
In the J-ALEX study, alectinib showed a better safety profile than crizotinib. The most common toxicity with alectinib was confirmed as being constipation (36%). Grade 3 or 4 AEs occurred at a greater frequency with crizotinib (52%, 54 of 104 patients) than alectinib (26%, 27 of 103 patients). Dose interruptions due to AEs were also more recurrent with crizotinib (74%, 77 of 104 patients) than with alectinib (29%, of 103 patients), and more patients receiving crizotinib (20%, 21) than alectinib (9%, 9) discontinued the study drug because of an AE. No fatal AEs were reported in either treatment group. 52
Finally, in the ALEX trial, anaemia (20%), myalgia (16%), increased blood bilirubin (15%), increased weight (10%), musculoskeletal pain (7%), and photosensitivity reaction (5%) occurred with increased incidence in the alectinib arm than in crizotinib one. AEs leading to alectinib dose reduction, interruption, or discontinuation were reported in 16%, 19% and 11% of subjects, respectively. 47 Considering previous data, among all AEs, the most important was hepatotoxicity, even if it was <5% for grade 3 or 4; it occurred often within the first 2 months of treatment initiation. Myalgia and CPK elevation were also present, while sinus bradycardia was unique to alectinib but occurred less frequently than musculoskeletal AEs. Alectinib was associated with low incidences of gastrointestinal AEs or lower incidences of interstitial lung disease. Finally, clinical trials of alectinib did not have, for now, sufficient number of geriatric patients to help determine whether or not dose adjustment was necessary for this population, neither to differentiate outcomes in this specific subgroup. 53 Gender, or body weight did not have a clinical meaningful impact on exposure to alectinib or its metabolite M4, and considering Asian versus non-Asian race, according to Mok and colleagues’ subgroup analysis, AE profiles were consistent with the intent-to-treat population (ITT) population. Diarrhoea was more common with crizotinib in both subgroups versus alectinib: Asian, 15% (with alectinib), 39.1% (with crizotinib); non-Asian, 10% (with alectinib), 50% (with crizotinib). Nausea was more common with crizotinib in both subgroups versus alectinib: Asian, 10.1% (with alectinib), 42% (with crizotinib); non-Asian, 17% (with alectinib), 52.4% (with crizotinib). Similar results were evident about rate of treatment discontinuation due to AEs: Asian 13% (with alectinib), 11.6% (with crizotinib); non-Asian 9.6% (with alectinib), 13.4% (with crizotinib).51,53
Alectinib: efficacy on brain metastases
About 8% of NSCLC patients present with brain metastases at diagnosis, while 25–30% of NSCLC patients will develop them during the course of their disease. Prognosis is poor, with a median OS of about 3 months. 19 In ALK-rearranged NSCLC subjects, crizotinib efficacy on CNS disease is limited by low BBB penetration. Conversely, alectinib is particularly effective against CNS metastasis.
Furthermore, early small data sets showed an intracranial response rate to alectinib ranging from 40% to 57% and, in the October 2016, Gadgeel and colleagues, in a pooled analysis from the two previously described single-arm phase II studies (NP28761 and NP28673), significantly clarified CNS response to the drug. 54 Secondary endpoints in both studies included CNS overall response rate (CORR), CNS disease control rate (CDCR), and CNS duration of response (CDOR). Of the overall study populations, 136 patients (60%) with CNS metastases at baseline were assessed for intracranial response. Fifty patients (37%) had measurable CNS disease at baseline. Ninety-five of them (70%) underwent prior CNS radiotherapy (55 patients had CNS radiotherapy more than 6 months prior to alectinib initiation). Median follow-up time was about 1 year. Brain scans were taken every 6 weeks in the NP28761 study and every 8 weeks in NP28673. For patients with baseline measurable CNS disease, CORR was 64% (95% CI, 49.2–77.1), CDCR was 90% (95% CI, 78.2–96.7%), with a median CDOR of 10.8 months (95% CI, 7.6–14.1 months). For those with measurable or nonmeasurable baseline CNS disease, CORR was 42.6% (95% CI, 34.2–51.4%), CDCR was 85.3% (95% CI, 78.2–90.8%) and median CDOR was 11.1 months (95% CI, 10.3 months to NE). When stratified by prior radiotherapy (pre-specified) responses were seen in 35.8% (95% CI, 26.2–46.3%) with prior radiotherapy (n = 95) and 58.5% (95% CI, 42.1–73.7%) without prior radiotherapy (n = 41). Complete intracranial responses were observed in 18% of patients with prior radiotherapy and 49% of those without prior radiotherapy, respectively.54–56 As authors stated, and considering Tran and colleagues’ comments, potential weaknesses of the analysis included a small sample size and the single-arm design of the two studies, which did not provide CSF alectinib concentrations as previously done by Costa and colleagues in their case report. 57
In the ALUR study, CORR was a secondary outcome: in patients with measurable disease, it was 54.2% in the alectinib arm versus 0% in the chemotherapy one (difference 54.2%, 95% CI, 0.23–0.78). 45
Results from the frontline approach in J-ALEX and ALEX trials provided further evidence of alectinib’s systemic and CNS efficacy, with complete CNS responses rates of 38% in patients with measurable CNS lesions at baseline. In the ALEX ITT population, the cumulative incidence rate (CIR) of CNS progression, considering the challenging risks of non-CNS progression and death, was 9.4% with alectinib versus 41.4% with crizotinib. When they were analysed according to baseline CNS metastases condition, CIR trends observed for CNS progressive disease (PD) versus non-CNS PD in the ALEX trial were similar to those in Gadgeel and colleagues’ pooled analysis described above, confirming alectinib’s effectiveness in preventing or delaying CNS metastases in ALK-positive NSCLC. 58
If, ultimately, a CNS PD occurs under alectinib administration, dosing strategies to overcome reduced CNS activity have also been explored, since Gainor and colleagues recently reported that alectinib dose escalation (900 mg twice daily) reinduced CNS tumour response in two patients with ALK-positive NSCLC, who experienced CNS relapse on standard-dose alectinib (600 mg twice daily). 59
Alectinib: place in therapy
Alectinib, as demonstrated by the ALEX trial, is a more suitable ALK inhibitor than crizotinib as standard first-line therapy for ALK-positive lung cancer in terms of efficacy, toxicity, and prevention of CNS metastases, but crucial issues still need addressing.
Among critical concerns are that molecular data need to be highlighted: different EML4–ALK fusion variants predicted differential response and disease control to crizotinib; this is because protein stability of EML4–ALK variants influences the overall fusion protein stability, inhibitor-induced protein degradation and drug sensitivity. In particular, it was evident that patients with EML4–ALK variant 1 had similar ORR to crizotinib (74% versus 63%) but higher disease control rate (95% versus 63%) and longer median PFS (11 versus 4.2 months) than individuals with other variants. 60 Woo and colleagues showed in their study a 2-year PFS of 69% (95% CI, 49.9–95.4) in group variants 1/2/others versus 32.7% (95% CI, 15.6–68.4) in group variants 3a/b (p = 0.108) among all crizotinib-, alectinib-, and ceritinib-treated patients. Variant 3a- or 5a-harbouring cells were resistant to ALK inhibitors with >10-fold higher half maximal inhibitory concentration in vitro. Considering these conditions, the most efficacious ALK inhibitor is still unknown. 61 Moreover, clinical data showed that particular subpopulations were less represented in clinical studies: in the ALEX trial, only 20 (6.6%) patients were classified as Eastern Cooperative Oncology Group (ECOG) performance status = 2, and only 17 (5.6%) patients were active smokers; in those cases, alectinib presented uncertain outcomes. 9
With the aim of defining an accurate algorithm for this population of patients, it is noteworthy to consider that if the usual PFS on first-line crizotinib, as evidenced in the PROFILE 1014 trial, is around 11 months, and that of alectinib, at time of progression on crizotinib, is around 8–9 months, the purpose of a second-generation frontline agent exceeding 19 months should be considered superior to the sequential approach. 62 At present, PFS with alectinib, as first-line treatment, is clearly superior (25.7 months according to updated results of the ALEX trial, doubling the 11 months of crizotinib arm in the same trial) to combined PFS of crizotinib followed by alectinib, with an incidence of brain metastases much lower (9% versus 41%, respectively). Alectinib upfront in ALK-positive metastatic patients performed better also in comparison with crizotinib followed by ceritinib (PROFILE 1014/ASCEND3), ceritinib alone (ASCEND4) or crizotinib followed by brigatinib (PROFILE 1014/phase I-II trial) and, even if these comparisons are only hypothetical, considering also efficacy and safety, alectinib should be a considerable choice in the first-line approach. With the aim of prolonging PFS and finally OS, further indications will also arrive from comparisons among alectinib and new ALK inhibitors such as lorlatinib or ensartinib, which are still under evaluation. Repeated biopsies will play a larger role in guiding decisions about the best approaches or overcoming resistance in order to improve survival for this small subgroup of patients. 62
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
Dr Tiziana Vavalà declares no conflicts of interests. Professor Silvia Novello declares these conflicts of interests in the last 3 years: speaker bureau Eli Lilly, BMS, BI, Incyte, MSD, Roche and Astra Zeneca.