
Abstract
Cervical cancer is the fourth most common malignancy among women worldwide, and continued research to discover biomarkers or therapeutic targets will aid early diagnosis and treatment of this cancer. Here, we investigated novel cervical cancer biomarkers using integrated analysis of high-throughput sequencing data from the Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) databases. We have identified nine genes of interest that appear to be involved in cervical cancer development: SPARCL1, SYCP2, KIF4A, PRC1, TOP2A, LAMP3, KIF20A, MCM2, and APOBEC3B. Furthermore, gene ontology (GO) and co-expression analysis of these differentially expressed genes indicated that SPARCL1 may play a core role in cervical cancer development. Further, we analyzed the expression of these nine genes during the progression of cervical cancer, and found that SPARCL1 is also related to precancerous lesions and migration processes during cervical cancer pathogenesis. Finally, we validated these observations by investigating SPARCL1 expression in cervical cancer tissue and serum samples. The diagnostic specificity of serum SPARCL1 in cervical cancer occurrence was also compared with other high incidence diseases. All of these data indicate that SPARCL1 may be a novel cancer predictive marker and a potential therapeutic target for tumor development and progression in cervical cancer.
Keywords
Introduction
Cervical cancer is the second largest type of malignant tumor in women, second only to breast cancer, with more than 1.5 million new cases diagnosed each year and over 300,000 deaths [1, 2]. It has also been associated with persistent infections with oncogenic human papilloma viruses (HPVs). However, while HPV infection is an indispensable factor, it alone is not sufficient to cause cancer. In fact, the majority of acute HPV infections induce low grade precursor lesions that are cleared spontaneously after several months in more than 90% of cases, with less than 10% of cases eventually progressing to high grade lesions or invasive cancer [3]. Cervical intraepithelial neoplasia is the typical type of precancerous lesion leading to cervical cancer, and in the evolution from precancerous lesion to malignant tumor, the expression of a variety of oncogenes and invasion-related genes is significantly altered [4]. At present, the key molecules causing cervical intraepithelial neoplasia to develop into cervical cancer have not been elucidated [5]. Thus, the discovery of novel biomarkers that will allow these molecular events to be monitored in histological or cytological specimens is essential and will likely help improve the detection of high risk lesions in both primary screening and triage settings [6].
The Gene Expression Omnibus (GEO) repository at the National Center for Biotechnology Information (NCBI) archives and freely disseminates microarray and other forms of high-throughput data generated by the scientific community. The database has a minimum information about a microarray experiment (MIAME)-compliant infrastructure that includes fully annotated raw and processed data, which includes expression data for a variety of tumors [7, 8]. The Cancer Genome Atlas (TCGA) project has analyzed mRNA expression, microRNA expression, promoter methylation, and DNA copy number in 306 cervical squamous cell carcinomas and 3 normal cervical tissue samples [9, 10]. Thus, combining the GEO and TCGA datasets may provide important insight into novel biomarkers of these tumors. Indeed, screening of tumor biomarkers based on the GEO and TCGA data has recently revealed a series of highly specific and sensitive markers [11, 12]. Compared with conventional screening methods, analyzing high-throughput data based on bioinformatics methods allows researchers to obtain stable and reliable biomarkers in a larger number of clinical samples.
Meta-analysis is a statistical analysis method that combines the results of multiple scientific studies using a statistical approach in an effort to increase power, improve estimates of the size of the effect, and/or to resolve uncertainty when reports disagree [13, 14]. With regard to screening biomarkers, combining multiple GEO datasets can help to validate the expression of multiple genes of interest between normal and tumor tissues, analyze the specificity and sensitivity of a biomarkers, and even correlate the expression of genes of interest with patients prognosis [15, 16].
Here, we analyzed three independent datasets from the GEO database and TCGA-CSCC, which contains normal cervical and cervical cancer samples. In doing so, we identified nine-gene signatures that were associated with cervical cancer occurrence: SPARCL1, SYCP2, KIF4A, PRC1, TOP2A, LAMP3, KIF20A, MCM2, and APOBEC3B. Differences in gene expression were then evaluated using meta-analysis, which further proved that these nine genes are indeed related to cervical cancer pathogenesis. Diagnostic meta-analysis also indicated that the nine gene signatures have prominent specificity and sensitivity in cervical cancer diagnosis. GO analysis and co-expression analysis showed that SPARCL1 may be a core factor in the development of cervical cancer. Subsequently, we analyzed the expression of those nine-gene signatures in the progression of cervical cancer and found that SPARCL1 is also related to precancerous lesions and the migration process of cervical cancer. Finally, we validated the correlation of SPARCL1 in cervical cancer development and progression in cervical cancer tissue and serum samples. These data indicate that SPARCL1 may be a novel predictive marker and therapy target for tumor occurrence and progression in cervical cancer.
Materials and methods
Materials
Cervical cancer tissue microarrays (CR602a and CR806) were purchased from Biomax US (MD, USA). The anti-SPARCL1 polyclonal antibodies were obtained from ProteinTech Group, Inc. (Wuhan, China). The human SPARCL1 ELISA Kit (EME-H8276) was purchased from KITZOME (Shenzhen, China). All other kits or reagents were purchased from the Beyotime Institute of Biotechnology (Shanghai, China).
Differential gene screening
To identify relevant studies, we obtained sequencing data from TCGA database. We also retrieved all relevant studies from the GEO database. The dataset search was limited to those in the English language and that were completed in or before October 2016. To increase the sensitivity of the search, both mesh-terms and free words were used. The search terms included: “cervical cancer”; “normal”; and “tissue” or “tumor” or “cancer” or “carcinoma” or “neoplasm”.
Three GEO datasets (GSE6791, GSE7803, GSE390 01) which contain normal cervical tissue as well as cervical cancer tissue were downloaded. The “Series Matrix Files” and their corresponding platform files were integrated by Perl language to create a new generated file containing gene symbols and their corresponding expression in each sample. We then analyzed differential gene expression in the normal cervical group and cervical cancer group by R language based on the limma library. These data were used to generate the corresponding heatmap, volcano figure, and table of differentially expressed genes.
Files from TCGA-CSCC project were downloaded from TCGA database. The download terms included “Transcriptome Profiling” and “Gene Expression Qua- ntification” for the Data Category and “RNA-Seq” and “HTSeq-Counts” for the Experimental Strategy and Workflow Type, respectively. The analysis included 306 cervical squamous cell carcinoma and 3 normal cervical tissue samples. Similar to our analysis above, the files were integrated with Perl language to create a new generated file containing the gene symbols and their corresponding expression in each sample. We then analyzed the differences in gene expression in the normal and cervical cancer groups by R language based on the edgeR library. These data were then used to generate the corresponding heatmap, volcano figure, and differential gene expression table. The genes identified in both our GEO and TCGA database analyses are presented with a Venn-diagram.
Data extraction and meta-analysis
Data extraction was repeated independently by three investigators, and a consensus was reached for each dataset. For the seven datasets available in the GEO database that included the expression and related patient data and sample characteristics (e.g., normal or cervical cancer tissue, TNM stage) were extracted for each of the nine genes of interest. We then calculated the expression of the nine genes in normal or cervical cancer tissue using GraphPad Prism 5. We analyzed the obtained data for the Mean, SD, and Total using Review Manager 5.3. An I
We also undertook a bivariate diagnostic meta-analysis to obtain pooled estimates of sensitivity and specificity, positive and negative likelihood ratios, and a summary diagnostic odds ratio. Summary Receiver Operator Characteristic (sROC) curves were then constructed using the bivariate model to produce a 95% confidence ellipse within the ROC space. Each data point in the summary ROC space represents a separate study.
GO enrichment analysis
The nine differentially expressed genes were also analyzed for their GO term enrichment using the Gene Ontology Enrichment Analysis Software Toolkit (GOEAST). The results of this GO analysis are grouped by Biological Process, Cellular Component, and Molecular Function.
Co-expression network using TCGA data
The nine genes of interests (SPARCL1, SYCP2, KIF4A, PRC1, TOP2A, LAMP3, KIF20A, MCM2, and APOBEC3B) discovered in our TCGA screening were analyzed for their co-expression and network connections. Briefly, all of the gene expression values were merged into one file. The weight of their relative coefficients were calculated and extracted by R language based on the WGCNA library using a threshold value of 0.80. The co-expression network was visualized by Cytoscape (version 3.4.0).
Immunohistochemical staining and assessment of SPARCL1 expression
Tissues microarrays were dewaxed and antigens were retrieved using high pressure for 3 min. Then, the activity of endogenous peroxidases was blocked using 3% hydrogen peroxide for 10 min at room temperature. After immersion in normal goat serum for 30 min at 37
The immunohistochemical staining was evaluated using a semi-quantitative scoring method. The scores were determined independently by two pathologists. SPARCL1 staining was scored according to staining intensity using the following grading system: no staining
Integrated analysis reveals nine genes of interest with altered expression during cervical cancer development. A. Venn-diagram presenting 72 distinct genes with a fold change greater than 2 between the four datasets investigated. B. Venn-diagram presenting 72 distinct genes with a fold change greater than 10 between the four datasets investigated. C–F. The expression of the nine differentially expressed genes displayed in clustering heatmaps. G–I. Top ten GO terms of the nine genes of interest based on their biological process (G), cellular component (H), and molecular function (I).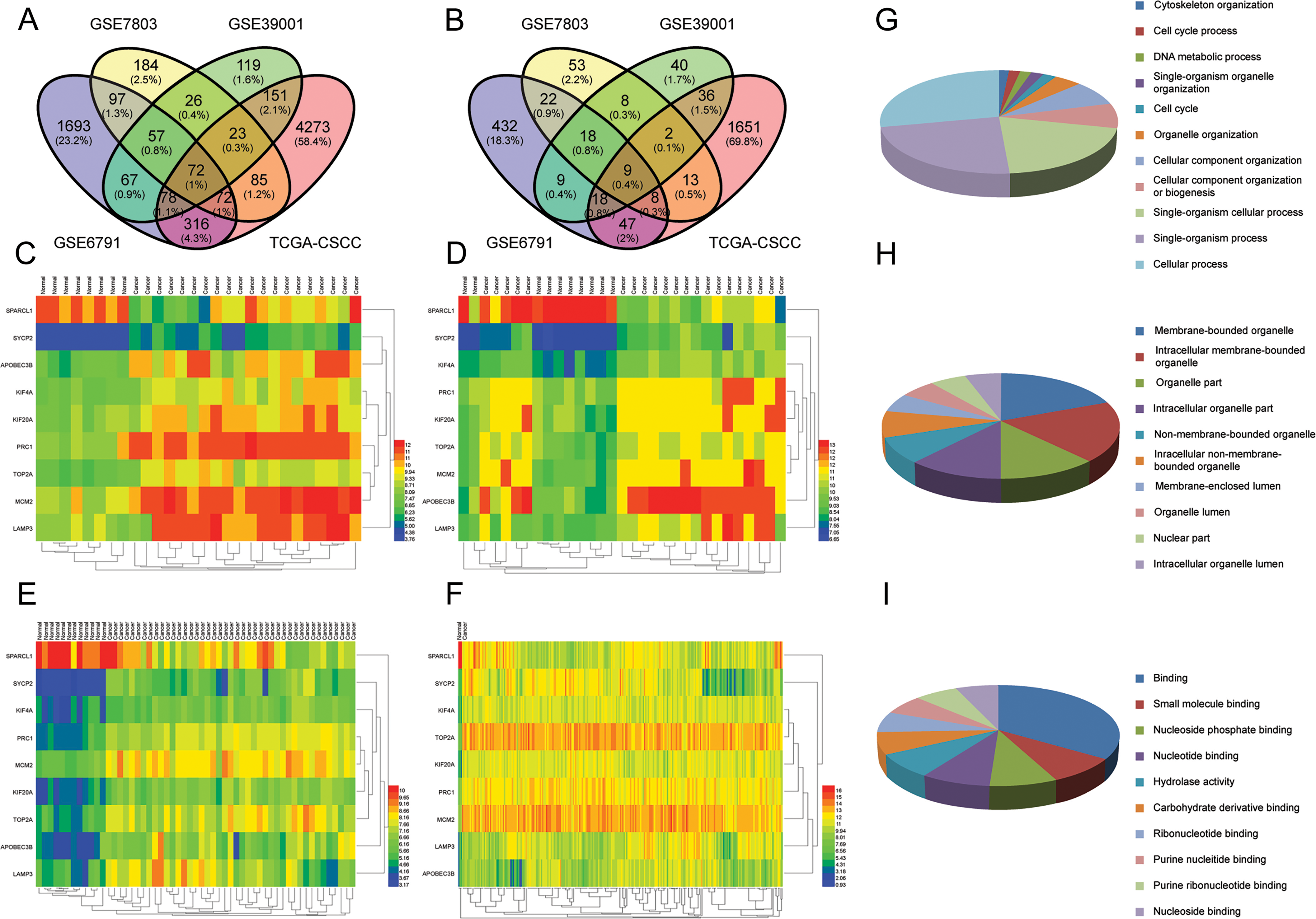
The serum samples collected and experiments conducted in this study were approved by the Chengdu Medical College ethics committee (No: CYYFYEC20 16002) in accordance with approved guidelines. Blood was collected prior to the first surgery and before patients receiving any kind of treatment. The collected serum samples were aliquoted into smaller volumes and stored at
SPARCL1 levels in the serum were measured and quantitated using an ELISA kit (KITZOME, Shenzhen, China) according to the manufacturer’s instructions. Each serum sample was analyzed three times.
Statistical analysis
Experiments were performed independently at least three times. One-way analysis of variance and two-tailed t tests were used to compare the effects of various treatments with GraphPad Prism 5 (San Diego, CA). Data are shown as mean
Study information on eight GEO datasets which consist of normal cervical and cervical cancer samples
Study information on eight GEO datasets which consist of normal cervical and cervical cancer samples
Meta-analysis of SPARCL1 expression in normal cervical and cervical cancer tissue. A. Meta-analysis of SPARCL1 expression in normal cervical and cervical cancer samples. B. Sensitivity and specificity of SPARCL1 in cervical cancer diagnosis. C. ROC curve of SPARCL1 as a diagnostic marker of cervical cancer development. D. The expression of SPARCL1 increases (
TOP 10 GO terms in each category (Biological process, Cellular component, and Molecular function) associated with the nine genes of interest analyzed by GOEAST
Clinicopathological association of SPARCL1 with cervical cancer
Clinical and TNM staging were referenced to American Joint Committee on Cancer: AJCC Cancer Staging Manual, 6th edition.
Integrated analysis reveals nine differentially expressed gene signatures that are associated with cervical cancer development
By integrating the four unique data sets (three GEO data sets (GSE6791, GSE7803, GSE39001) and TCGA-CSCC data), which contain normal cervical tissue and cervical cancer tissue, we created a Venn-diagram presenting 72 of the same distinct genes that changed greater than 2-fold. This analysis highlighted nine genes whose expression was altered more than 10-fold (Fig. 1A and B). In Fig. 1C–F, the expression of these nine genes in the four datasets is displayed in clustering heatmaps. Furthermore, our GO enrichment analysis of the nine gene signatures indicates a range of functions as highlighted by the list of top ten GO terms for their biological process, cellular component, and molecular function (Fig. 1G and I and Table 1). The full list of GO terms for these nine genes of interest can be found in Supplemental Figs 1–3.
Meta-analysis of expression level and diagnostic value of the nine genes of interest during cervical cancer development
While searching the GEO database, we excluded invalid datasets by reading their summary and sample characteristics (Supplemental Fig. 4). We included seven GEO datasets in this study (Table 2). Meta-analysis of their mean expression changes indicates that of the nine gene differentially expressed in cervical cancer samples compared to normal tissue (SPARCL1, SYCP2, KIF4A, PRC1, TOP2A, LAMP3, KIF20A, MCM2, and APOBEC3B), SPARCL1 appears to be more highly expressed in normal cervical tissue than in cervical cancer tissue (Fig. 2A and Supplemental Fig. 5). Additional diagnostic meta-analysis revealed that these nine gene signatures all have excellent diagnostic potential, are highly sensitive, and are specific for cervical cancer diagnosis (Fig. 2B and C and Supplemental Figs 6 and 7).
Progression-related expression and co-expression networks reveal SPARCL1 as an important factor in both cervical cancer development and progression
We analyzed the expression of the nine genes of interest in both precancerous lesions and later stage migration processes. In doing so, we found that the expression of SYCP2, KIF4A, PRC1, TOP2A, KIF20A, MCM2, and APOBEC3B was significantly increased (
Clinical significance of the expression of SPARCL1 in cervical cancer tissue samples and patient serum. A. Expression of SPARCL1 is related with clinical stage (
Diagnostic specificity of serum SPARCL1 expression in cervical cancer development. A. SPARCL1 is downregulated in cervical cancer, lung cancer, colon cancer, liver cancer, ovarian cancer, and breast cancer, while no changes were observed in prostate cancer, stomach cancer, or esophageal cancer. B. Expression of SPARCL1 is not associated with senile disease. C. Expression of SPARCL1 is not correlated with other chronic diseases.
A co-expression network of nine gene signatures also highlighted SPARCL1 as an important factor as it interacts with multiple other differentially expressed genes(Supplemental Fig. 10).
To determine the clinical significance of SPARCL1 expression in patients with cervical cancer, we performed immunohistochemical (IHC) labeling of specimens from a cohort of 102 patients with cervical squamous cell carcinoma, 10 samples of corresponding adjacent cervix tissue, 10 samples of chronic cervicitis, 10 samples of cervical intraepithelial neoplasia, and 8 samples of lymph node metastatic squamous cell carcinoma from the uterine cervix. SPARCL1 expression levels were measured with a semi-quantitative scoring method. Overall, SPARCL1 expression was observed in all patients and was significantly associated with clinical T classification (
During cervical cancer progression, SPARCL1 expr- ession was observed to significantly decrease (Fig. 3C). We further analyzed SPARCL1 expression in normal and cervical cancer patient serum. We found that serum SPARCL1 was downregulated in cervical cancer compared to normal tissue (Fig. 3D). Taken together, these data corroborate our conclusions based on our meta-analysis.
Diagnostic specificity of serum SPARCL1 in cervical cancer development
We tested serum SPARCL1 in healthy, high risk patients previously diagnosed with tumors (cervical cancer, lung cancer, colon cancer, liver cancer, prostate cancer, stomach cancer, ovarian cancer, esophageal cancer, and/or breast cancer), senile disease (diabetes, hypertension, atherosclerosis), and other chronic diseases (chronic hepatitis B, chronic obstructive pulmonary, chronic bronchitis, chronic renal failure). We found that SPARCL1 is downregulated in patients with cervical cancer, lung cancer, colon cancer, liver cancer, ovarian cancer, and breast cancer, while showing little to no change in prostate cancer, stomach cancer, esophageal cancer (Fig. 4A), senile disease (Fig. 4B), or chronic diseases (Fig. 4C).
Discussion
In recent years, with the implementation of the human genome project, widespread using of sequencing and gene chips has increased. This increase has also been further supplemented by the accessibility of multiple open databases, such as TCGA, GEO and Oncomine, which store vast amounts of gene expression data for both normal and diseased tissues [18, 19]. Integrating multiple studies from these databases allows researchers to investigate novel tumor biomarkers that have important research value [20, 21]. Based on the high morbidity and mortality rates associated with cervical cancer in gynecological tumors, we used an integrated analysis to evaluate novel biomarkers in cervical cancer in multiple TCGA and GEO data sets. Gene screening demonstrated that nine genes of interest were differentially expressed (SPARCL1, SYCP2, KIF4A, PRC1, TOP2A, LAMP3, KIF20A, MCM2, and APOBEC3B) and are likely related to cervical cancer development and progression.
Meta-analysis as a systemic method for evaluating multiple studies [14]. In the present study, the nine differentially expressed genes were further analyzed in terms of their specificity and sensitivity as cervical cancer biomarkers using both differential and diagnostic meta-analysis. In doing so, we found that the nine gene signatures were significantly different in normal and cervical cancer tissues. Thus, the expression of the nine genes can indeed be used as diagnostic biomarkers of cervical cancer.
Of the nine genes of interest identified in this study, SYCP2 has been previously related to HPV-associated cancer, screening, and development of cervical cancer [22, 23]. The expression and prognostic significance of KIF4A [24, 25], TOP2A [26, 27], LAMP3 [28, 29], KIF20A [30, 31], and MCM2 [32, 33] have also been reported in a wide variety of tumor types, including a Phase I/II clinical trial of a peptide vaccine derived from KIF20A in patients with advanced pancreatic cancer [31]. Moreover, APOBEC3B appears to be related with HPV, hepatitis virus B (HBV), and associated cancers [34, 35]. Thus, it seems likely that these genes are related to cervical cancer and that our biomarker screening methods are valid, resulting in a list of valuable differentially expressed genes that have application potential.
Progression-related expression and co-expression networks also revealed that SPARCL1 may play an important role in both cervical cancer occurrence and progression. Our analysis revealed that this particular gene may be involved in the expression changes observed in the other eight genes, making it a particularly important and interesting biomarker. Recent studies have shown that SPARCL1 is down-regulated in a number of cancers, including colorectal cancer [36], gastric cancer [37], breast cancer [38], prostate cancer [39], hepatocellular carcinoma, and non-small cell lung cancer [40]. However, no studies have been conducted concerning its clinical significance and diagnostic value in cervical cancer. Thus, we analyzed the expression of SPARCL1 during cervical cancer progression by tissue array and found that SPARCL1 expression is also down-regulated.
Currently, the biggest limitation for the clinical application of tumor markers is poor specificity [41]. Most tumor biomarker research has focused on differences in normal and tumor tissue or serum in patients, while ignoring research on the interference expression of these candidate biomarkers in other common diseases [42, 43], including cardiovascular disease and chronic diseases. Thus, evaluating the expression of potential cancer biomarkers in multiple diseases can provide a new method for investigating their specificities. In this study, we found that SPARCL1 is specifically regulated in a wide variety of tumors, while have no obvious changes in other senile or chronic diseases compared with normal samples. Taken together, our data indicates that SPARCL1 may be an excellent tumor biomarker and its clinical application warrants further investigation.
Footnotes
Acknowledgments
This work was funded by the Foundation of Sichuan Province Science and Technology Agency (2014JY00 39), Foundation of Sichuan Province Education Office (15ZB0245), Foundation of Sichuan Medicine Institute (16PJ112), and Foundation of Chengdu Medical College (CYTD15-03).
Conflict of interest
The authors have no competing financial interests to disclose.
Supplementary data
The supplementary files are available to download from