
Abstract
BACKGROUND:
Pancreatic adenocarcinoma (PDAC) is one of the deadliest human malignancies. Although surgery is currently the only effective treatment for PDAC, most patients survive less than 20 months after tumor resection.
OBJECTIVE:
The primary goal was to investigate alterations in KRAS, TP53, SMAD4 and CDKN2A/p16 in tumors from patients with exceptionally long survival after surgery.
METHODS:
Tumors from 15 patients with PDAC that survived more than 55 months after surgery (“LS”) were analyzed for KRAS, TP53, IDH1, NRAS and BRAF using next-generation sequencing. SMAD4 and CDKN2A/p16 was tested using immunohistochemistry. MGMT promoter methylation was investigated.
RESULTS:
Tumors from “LS” have a lower prevalence of KRAS and TP53 mutations and had more frequently SMAD4 retained expression, if compared with that of patients died within 24 months from surgery. The survival of patients with wild-type KRAS and TP53 tumors was more than twice longer than that of patients bearing KRAS and TP53 mutations (90.2 vs. 41.1 months). Patients with KRAS wild-type tumors and that retained SMAD4 expression had a survival twice longer than cases with alterations in both genes (83.8 vs. 36.7 months). Eleven tumors (39.3%) showed MGMT methylation.
CONCLUSIONS:
Our data indicate that absence of KRAS, TP53 and SMAD4 genetic alterations may identify a subset of pancreatic carcinomas with better outcome.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest solid human malignancies [39], with overall 5-year survival well under 5% across all stages. Such a dismal prognosis is due to diagnosis at late stages, together with the lack of effective systemic therapies to control the disease [28]. Less than 20% of patients are amenable to surgery. Of those who undergo surgery, approximately 20% survive 5 years and only 10% are still alive after 10 years [11].
Surgical resection of PDACs offers the best hope to improve survival, but despite advances in pancreatic surgery, resected patients have a median survival
The factors responsible for long-term survival of patients with PDAC are poorly understood [11]. Numerous studies focusing on 5-year and 10-year survivors have shown that conventional clinicopathological parameters such as early stage of disease, lack of lymph node spread and negative surgical margins predict a more favorable prognosis [1, 18, 19, 26, 47]. However, these same studies also showed that positive resection margins or tumor metastases to lymph nodes did not preclude long-term survival. In fact, 20–40% of patients who survive more than 5 years after surgery had nodal disease and/or positive margins. These findings suggest that pathologic staging is not the sole determinant of a relatively long survival in patients with PDAC. The longer survival in this subset of PDAC patients may depend on distinct genetic, epigenetic, or other biologic factors, such as changes in the tumor microenvironment or enhanced immune response. The possibility of identifying those molecular parameters that can predict survival in the pre-operative setting would be most helpful for patient management and clinical decision making.
Researches in the field have uncovered genetic aberrations that occur during PDAC development and progression. In most cases, PDAC is initiated by oncogenic mutant KRAS [17]. Whole-exome sequencing of 24 PDACs was reported in 2008 and the exon analysis of 20,661 genes indicated that PDACs contain an average of 63 genomic alterations, most of which are point mutations [29]. High throughput next generation sequencing, including a large study by Bailey et al. [4], have shown that four main genes (referred to as “mountain genes” for their relevance) are altered in PDAC: KRAS, TP53, SMAD4, CDKN2A/p16 [21, 51, 58]. However, it is still under debate if there are molecular markers capable of predicting prognosis of PDAC patients [11, 45]. At present, medical societies exclude a need for personalized, molecularly targeted treatment of PDAC patients [15].
In the database of the Surgery Unit (Bologna Italy) we have identified 15 patients out of total 342 operated for PDAC who survived longer than 4 years after surgery (4.4%).
The aim of the present study is to define the molecular status for the KRAS, TP53, CDKN2A and SMAD4 “mountain genes” and of other potentially relevant markers such as IDH1, microsatellite instability (MSI) and MGMT (O6-methylguanine methyl-transferase) promoter methylation in these 15 PDAC patients with an unusually long survival.
Material and methods
Case selection
In this study, 15 patients with PDAC and a survival rate higher than 55 months from surgery (“long-survivor group” – LS) were analyzed.
All histologic diagnoses were reviewed by one of the authors (AF). Diagnosis, pTNM and stage evaluation were performed according to the World Health Classification (WHO) and the American Joint Committee on Cancer (AJCC) criteria, respectively [8, 16].
All samples were formalin fixed-paraffin embedded (FFPE) and obtained from resection specimens. They were analyzed for KRAS (exons 2-3-4) and TP53 (exons 5-6-7-8) mutational status. In KRAS wild-type samples, NRAS (exons 2-3-4) and BRAF (exon 15) genes were analyzed. Microsatellite instability (MSI) and MGMT promoter methylation status were also determined. Protein expression of p16 and SMAD4 was evaluated by immunohistochemistry.
A group of 15 patients with PDAC that died from the disease within 24 months from surgery (“non-long-survivor group” – NLS) were also analyzed: this group was matched for age, sex, stage and margin status with the “LS” group.
Primers used for mutational analysis
Primers used for mutational analysis
All information regarding the human material was managed using anonymous numerical codes. All samples were handled in compliance with the Helsinki Declaration (
In order to assess the molecular status of the genes analyzed, DNA was extracted from FFPE material scraped under microscope guidance from three 10-
After careful review of all histology sections, FFPE blocks with the largest representative areas of viable tumor cells were selected for molecular and immunohistochemical analyses (see section below). The fraction of mutated neoplastic cells was calculated normalizing the mutated allele fraction for the proportion of neoplastic cells in the area subjected to molecular analysis, as previously described [55].
Sequencing was performed using the 454 GS-Junior next generation sequencer (Roche Diagnostic, Mann- heim, Germany), as previously described [13] (
MGMT promoter methylation analysis
Methylation analysis of MGMT promoter region was performed according to previously published protocols [42].
Microsatellite instability
MSI analysis was performed investigating 12 microsatellite regions, as previously described [41].
Antibody used for immunohistochemistry analysis
Antibody used for immunohistochemistry analysis
FFPE blocks with the largest representative areas of viable tumor cells were selected for immunohistochemical analysis (and molecular analysis, see section above). Immunohistochemistry (IHC) was performed on two consecutive 3
CDKN2A/p16 is an oncosuppressor gene that is inactivated by mutation, loss of heterozygosity, epigenetic changes [8, 50]. Its inactivation, resulting in the lack of protein expression, is a very common event in pancreatic cancers [8, 15]. There is a statistical correlation between lack of p16 immunoreactivity and gene inactivation [49, 50]. Even if this correlation is not perfect, IHC is routinely used because it is the most cost effective way to infer gene inactivation, regardless the underlying molecular mechanism(s) [8]. The expression of p16 was scored based on the percentage neoplastic cells showing strong nuclear immunoreactivity. Tumors with
Statistical analysis
Categorical variables were compared using the Fisher exact test. Continuous variables were compared using the unpaired t-test or Mann-Whitney test. A p-value
Kaplan Meier method and Cox regression analyses were used to evaluate the mutational status as predictor of survival in the study population in the multiple regression analysis. Statistical analyses were performed using GraphPad Prism 6.01 (GraphPad Software) and Stata/SE 14.2 statistical software.
Results
“Long-survivor” group characterization
The group was composed of 15 patients, 8 males (53.3%) and 7 females (46.7%), aged from 45 to 78 years (mean 62.3 ys). Follow-up time ranged from 55 to 133 months (mean 100.4 ms, median 105.0 ms) (Table 3). All patients but one (died from disease with lung metastases) were alive at the time of the last follow-up. Clinicopathological features and molecular status for each cohort patients are reported in Supplementary Table 1. Pancreatic ductal adenocarcinomas were classified as follows: 10 classic PDAC (two of them associated with Intraductal Papillary Mucinous Neoplasia – IPMN), and five morphological variants of PDAC (Supplementary Table 1). These latter were: one medullary carcinoma associated with IPMN, one colloid carcinoma, one undifferentiated carcinomas with osteoclast-like giant cells, one PDAC Large-gland type and one PDAC Large-duct type.
Clinical and molecular data of the samples analyzed compared with data reported in the literature
Clinical and molecular data of the samples analyzed compared with data reported in the literature
Twelve patients had a stage II disease (9 patients IIA and 3 patients IIB) and 3 a stage I disease (one patient IB, and 2 patients IA). The vast majority (12 of 15, 80.0%) were pT3 and had no lymph nodal involvement at presentation (Table 3). No distant metastases were observed at presentation in our cohort. Fourteen patients had negative resection margins (R0 disease), whereas 1 patient had positive margins (R1 disease) (Table 3).
In 9 patients (60.0%) a KRAS mutation was identified (Table 3). All the KRAS mutations were in exon 2. In the 6 patients wild-type for KRAS gene, no mutations in NRAS (exons 2-3-4) or in BRAF (exon 15) genes were detected (Table 3). Five patients (33.3%) harbored a TP53 mutation (Table 3). In 3 of these 5 patients also a KRAS mutation was observed (Table 3). No single nucleotide polymorphisms (SNPs) were detected in the amplicons analyzed.
To address tumor heterogeneity, we estimated the percentage of mutated neoplastic cells for KRAS or TP53 (see materials and methods): the mutated neoplastic cell range was 43–100% (mean 68.9%) and 71.4–88.9% (mean 81.5%), respectively.
Six of 14 evaluable samples (40.0%) were MGMT-methylated. In no one of the analyzed samples was detected MSI or mutation in IDH1 gene (Table 3).
In ten samples, the percentage of neoplastic cells positive for p16 were
Two samples were negative or scored as “1+” for SMAD4 staining, 11 were “2+” or “3+” and in two samples the staining was not evaluable (Table 3).
Clinicopathological and molecular features of the “NLS” group are described in Table 3 and Supplementary Table 1.
Clinicopathological features. The 15 “NLS” were matched with “LS” for age, sex, stage and margin positivity (Table 3). “LS” group showed less frequent lymph nodal involvement (20.0% vs 73.3%,
As expected, the survival time from surgery was significantly higher in the “LS” group (88.1 ms vs 9.7 ms,
Molecular status. The “LS” cases had a lower frequency of KRAS mutations (60.0% vs 86.7%), although the difference did not reach statistical significance (
To address tumor heterogeneity, we estimated the percentage of mutated neoplastic cells for KRAS or TP53 (see materials and methods): the mutated neoplastic cell range was 30–100% (mean 70.2%) and 30–100% (mean 64.9%), respectively.
No significant differences were observed in MGMT promoter methylation status between “LS” (42.9%) and “NLS” (33.3%) (
Overall survival according to the status of molecular markers
To evaluate if molecular markers predicted prognosis, we evaluated patient survival according to the molecular status of their tumors from the time surgery for all cases (15 “LS” and 15 “NLS”).
Patients with KRAS-WT carcinomas had a survival 1.5-fold higher than patients with tumors KRAS-mutated (77.1 ms vs 47.1 ms, respectively;
Survival analysis according to the status of molecular markers
Survival analysis according to the status of molecular markers
OS: Overall Survival; IHC: immunohistochemistry; WT: Wild-Type; mut: mutated; ms: months; pos: positive; neg: negative.
Survival of 30 PDAC patients (15 long surviving and 15 controls) according to the status of molecular markers (Kaplan-Meyer curves). A) KRAS mutated vs. wild type cases. B) TP53 mutated vs. wild type cases. C) SMAD4 loss-of-expression cases vs. cases with retained expression. D) 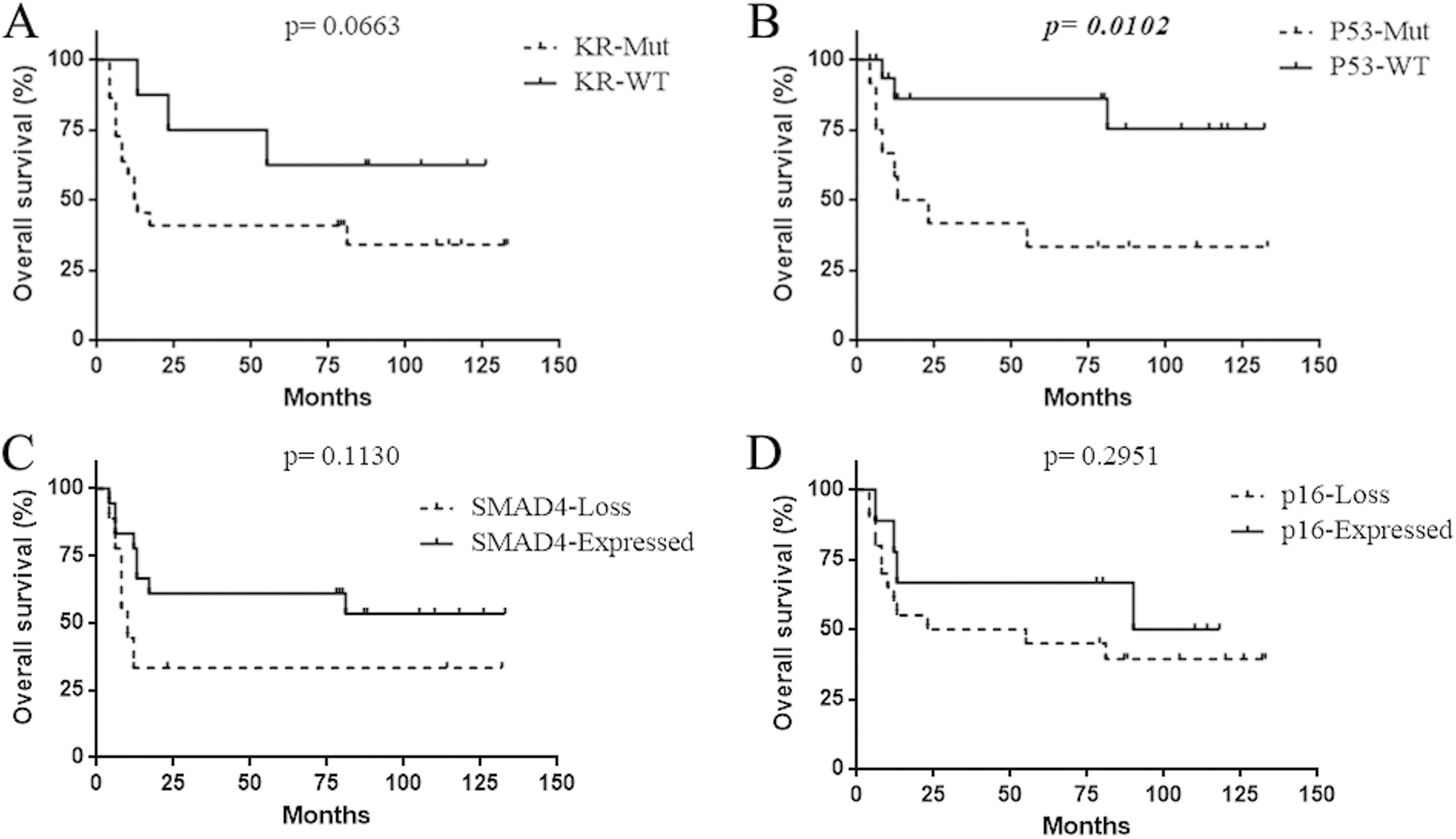
SMAD4 positive cases (scores 2+ and 3+) had a mean survival nearly double than that of SMAD4 negative ones (scores 0 and 1+) (56.4 ms vs 30.2 ms), but the difference did not reach statistical significance (
No difference in mean survival was also observed between MGMT promoter-methylated samples and those that did not show promoter methylation (59.9 ms vs 53.1 ms;
Survival of 30 PDAC patients (15 long surviving and 15 controls) according to the pattern of combined molecular alterations (Kaplan-Meyer curves). A) KRAS and TP53 mutated cases vs. cases wild-type for both. B) Mean survival of cases according to KRAS and TP53status. C) KRAS mutated and SMAD4 negative cases vs. cases KRAS wild-type and SMAD4-positive. D) Mean survival according to KRAS and SMAD4 status. E) TP53 mutated and SMAD4 negative cases vs. cases TP53 wild-type and SMAD4-positive. F) Mean survival according to TP53 and SMAD4 status. KR+TP53: KRAS and TP53 mutated cases; KR+SMAD4: KRAS mutated and SMAD4negative cases; KR: cases mutated only for KRAS; TP53: cases mutated only for TP53; WT/WT: cases wild-type for both gene sets analyzed (KRAS and TP53; KRAS and SMAD4).
Intriguingly, when we compared the survival of cases that were KRAS and TP53 mutated with that of cases that were wild type for both, we observed that the survival of the latter group was more than twice longer (90.2 ms vs 41.1 ms,
Patients with PDAC KRAS-WT and with positive expression of SMAD4 had a survival time twice longer than subjects with a PDAC KRAS-mutated and with negative expression of SMAD4 (83.8 months vs 36.7 months;
Patients with TP53-WT PDAC and positive expression of SMAD4 had a survival time five-times longer than subjects with a TP53-mutated PDAC and negative expression of SMAD4 (65.1 months vs 12.3 months;
The “LS” and the “NLS” group differed with regards to the presence of lymph node metastases that was statistically lower in the “LS” group (see above). For this reason, to further investigate the relationship of KRAS and TP53 mutations, and of SMAD4 loss of expression with survival we performed multivariate analysis. The hazard ratio (HR) for poor survival was 2.72 (95% confidence interval – CI: 0.33–22.2;
Too few cases lacked gene alterations in KRAS and TP53 and SMAD4 (triple negative cases KRAS-WT and TP53-WT and SMAD4-positive;
Alterations of KRAS, TP53, SMAD4 and CDKN2A/ p16 define the molecular profile of PDAC, but their prognostic and clinical implications are still controversial [5, 6, 22, 48, 52]. One of the reasons is the dismal prognosis of most patients with PDAC: the few operable patients often die from disease within a few months after surgery. The background of genetic alterations in the few patients that survive years after surgery is not well documented. In the present study, we have analyzed the status of the KRAS, TP53, p16 and SMAD4 in tumors from long survivors of PDAC (“LS” patients). We also investigated the molecular status of IDH1, microsatellite instability and MGMT promoter methylation, all of which have been reported with low prevalence in PDAC, reasoning that their prevalence may be higher in cases associated with an unusually long survival.
We showed that tumors from long PDAC survivors have the same general alterations found in those of patients that follow the usually dismal course, similar to what reported by Dal Molin et al. [11].
However, we found some important differences after comparison with a PDAC control group (“NLS” patients) from our institution and with the data reported in large series in the literature. Tumors of long PDAC survivors from our group had a remarkably lower prevalence of KRAS mutation and of SMAD4 loss of expression. We also found that absence of KRAS and TP53 mutations, and that absence of KRAS mutations combined with positive SMAD4 expression correlated with longer survival. We have observed that “LS” patients have a low frequency of KRAS mutations (60.0%) when compared with “NLS” patients (86.7%) and with data reported in literature (90%). Also TP53 mutations are less frequent in tumors from “LS” patients when compared to “NLS” PDAC patient controls and data from the literature, and the survival of cases with TP53 mutation was higher than that of the TP53-WT cases. Interestingly, the coexistence of KRAS and TP53 mutations was correlated with survival. Patients without KRAS and TP53 mutations had a survival rate more than twice longer when compared with subjects carrying tumors with KRAS and TP53 mutations. Furthermore, multivariate analysis showed that patients without KRAS and TP53 mutations had a lower risk of dying of disease compared with patients with KRAS and TP53 mutated tumors, regardless of lymph node metastases. The risk difference was statically significant in patients with lymph node involvement. These observations appears in line with experimental data showing how oncogenic KRAS species are not only involved with tumor development and progression [14], but also promote the development of adenocarcinoma from pancreatic ductal cells when TP53 is inactivated by mutation [3].
Regarding the CDKN2A/p16 status, no major differences were observed in p16 immunohistochemical reactivity between tumors from “LS” and “NLS” patients. On the other hand, SMAD4 was less frequently lost in tumors from “LS” (15.4%) compared with “NLS” patients and data from the literature (50% and 55% respectively). Moreover, absence of alterations in KRAS and SMAD4 seem to identify a subset of patients with a twice longer survival compared with cases harboring both KRAS mutation and SMAD4 loss of expression.
This data indicates that absence of KRAS and TP53 mutations and a preserved SMAD4 expression may identify patients with a favorable prognosis. Similar results have been recently observed in lung adenocarcinomas, where KRAS and TP53 mutations identify patients with worse clinical outcome [36].
We have also investigated whether MGMT promoter methylation, IDH1 mutations or microsatellite instability may be enriched in tumors from “LS” patients.
MGMT promoter hypermethylation confers sensitivity to alkylating agents (e.g. temozolomide) in patients with gliomas [27]. Contrary to what observed by Ueki et al. [53], but in accordance with Peng et al. [46], we observed that the MGMT promoter can be hypermethylated in PDAC. The higher percentage of MGMT promoter-methylated PDAC found in our cohort compared to that reported by Peng et al. may be due to the higher sensitivity of the method adopted in our study. This epigenetic modification has been observed both in the “LS” and “NLS” groups, but MGMT promoter methylation did not influence survival. Nevertheless, MGMT promoter methylation may be considered as a predictive marker for therapy with temozolamide as in the case of gliomas. The treatment of advanced pancreatic cancer with temozolamide has been attempted in a phase II study in 1998 [40], but no clinical response was seen in the 15 patients subjected to treatment. It is worth noting that, in the study, the treatment was performed without selecting patients according to MGMT promoter methylation status. Efficacy of alkylating agents has been demonstrated in neuroendocrine forms of pancreatic cancer [20].
Somatic mutations in the isocitrate dehydrogenase 1 gene (IDH1) have been described in acute myelogenous leukemia, cartilaginous tumors and gliomas [10], and have been linked to epigenetic deregulation that contributes to tumorigenesis [31, 35]. Few studies have investigated IDH1 in PDAC. In our series, no exon 4 IDH1 mutations were identified, a result compatible with the very low prevalence of mutations found in other studies [7, 9].
The risk of developing PDAC is increased in the Lynch syndrome caused by germ-line mutations in mismatch repair genes [25]. Tumors with mismatch repair defects typically feature a high degree of microsatellite instability (MSI) [30, 33]. The MSI-High phenotype is frequent in colorectal cancers, mainly in mucinous types arising in the right side of the colon and has been associated with a relatively favorable prognosis. Most pancreatic adenocarcinomas have a MSI-stable or sometimes a MSI-Low phenotype [2, 23, 24, 32, 37]. Only the few pancreatic adenocarcinomas occurring in the setting of Lynch syndrome and/or exhibiting poorly differentiated medullary histopathology display the MSI-High phenotype [2, 32, 37, 57, 59]. All tumors analyzed from our “LS” patients were MSI-stable, as were those from “NLS” controls. None of our cases was associated with Lynch syndrome or showed poorly differentiated medullary features. Thus, our data do not support a correlation between the MSI-High phenotype and prognosis, at variance with what reported by Nakata and colleagues [43].
Various clinical factors such as tumor size and differentiation, lymphatic and/or vascular invasion, tumor stage, involvement of resection margins influence outcome of PDAC patients. However, these parameters cannot be evaluated (or can only be partially defined) before surgery is performed and the tumor removed. Alterations in KRAS, TP53 and SMAD4 can easily be assessed in the specimens used to establish a pre-operative diagnosis of adenocarcinoma (e.g. EUS-FNA and cytology samples) [13]. For mutation detection, we used a highly sensitive NGS protocol that requires a limited amount of input DNA and outperforms Sanger sequencing or commercial kits [12, 13]. In addition, NGS allows to evaluate the proportion of mutated alleles, an important piece of information to define a mutation as sub-clonal. Genetic heterogeneity has been reported in several tumors as well as in pancreatic cancers [34, 56]. Considering that pre-operative samples contain only a small part of the lesion, molecular heterogeneity is a well-known general limitation for the analysis of limited pre-operative samples. In our work, we addressed this problem by estimating the proportion of neoplastic cells carrying specific mutations (KRAS, TP53) and did not find genetic heterogeneity a relevant issue.
Applying a highly sensitive NGS protocol to pre-surgical samples may provide important information that the surgeon can integrate with the endoscopic evaluation and the pathological diagnosis to establish the best clinical management for each patient.
This cohort of patients is limited. Even if “LS” patients and a “NLS” control group are not clearly identified by specific pattern of mutations, the absence of KRAS, TP53 and SMAD4 alterations did identify in our study a subset of tumors with more favorable outcome. Considering how long survival of patients with pancreatic adenocarcinoma is exceptional, the relatively low prevalence of KRAS, TP53 and SMAD4 alterations in tumors from these patients definitely deserves additional investigation.
Footnotes
Acknowledgments
This work was supported in part by an Italian Government-Ministero Della Salute Grant No. RF-2011-02350857 to G.T.
Conflict of interest
The authors have nothing to disclose.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-170464.