
Abstract
OBJECTIVE:
The transcription factor FOXF2 is reported to be down-regulated in HCC. Its deficiency is correlated with shorter disease-free survival and overall survival of HCC patients; however, the mechanism remains to be elucidated.
MATERIALS AND METHODS:
In this study, we performed qRT-PCR and western blotting to confirm the down-regulated FOXF2 in HCC tissue and cell lines. Then the HCC cell line Huh7 transduced with FOXF2 shRNA was adopted in a series of in vitro and in vivo assays to evaluate the cell phenotype change, migration, invasion, proliferation, colonization of circulating cell and the formation of metastatic nodules.
RESULTS:
We found that FOXF2 was down-regulated in HCC tissues and cell lines. FOXF2 deficiency in Huh7 cells increased E-cadherin and decreased Vimentin. The down-regulation of FOXF2 impeded HCC cell migration and invasion capacity, but promoted the proliferation of HCC cells and the growth of subcutaneous tumors in nude mice, which indicated a mesenchymal-to-epithelial phenotypic change in Huh7 cells. FOXF2 deficiency enhanced the colonization of circulating HCC cell, thus promoted the formation of metastatic nodules.
CONCLUSIONS:
FOXF2 deficiency induced mesenchymal-epithelial transition (MET) in Huh7 cell which might facilitate the colonization of circulating tumor cells and the formation of metastasis.
Keywords
Introduction
Hepatocellular carcinoma (HCC) is the third leading cause of cancer-related death worldwide, and its incidence is increasing globally [1]. Despite the advances in the diagnostic methods, surgical equipment & techniques and multidisciplinary therapy, the prognosis of HCC patients remains poor, with a total 5-year survival rate of around 12% [2]. The metastasis and recurrence of HCC are obstacles in the way towards better outcomes [3], thus the study on the mechanisms of HCC metastasis and recurrence is in urgent demand.
The forkhead box transcription factor family is characterized by a highly conserved forkhead/winged helix DNA binding domain and functions in a variety of biological processes, like embryogenesis, tissue development, metabolism, differentiation, proliferation and apoptosis [4, 5]. The FOX family is also reported to have an important role in the progression of many cancer types and is correlated with their prognosis [6].
FOXF2 is a member of FOX superfamily and plays a part in tissue development, extracellular matrix metabolism and epithelial-mesenchymal interactions [7, 8]. Studies have revealed that decreased FOXF2 in some cancer types, such as breast cancer [9], esophageal squamous cell carcinoma [10], prostate cancer [11] and stage I non-small cell lung cancer (NSCLC) [12] , predicts poor prognosis of patients. Low FOXF2 level could predict shorter disease-free survival, shorter overall survival and a higher frequency of lymph node metastasis of certain cancer types. The mechanisms beneath the correlation between decreased FOXF2 and early-onset of metastasis and relapse of tumor have not been well understood. The agreement lies in the fact that FOXF2 deficiency promotes tumor cell proliferation and inhibits apoptosis; however, more researches are in need to reveal the possible mechanisms. Wang et al. found that FOXF2 deficiency in basal-like breast cancer activated epithelial-mesenchymal transition (EMT) through up-regulating TWIST1 [13]. The research by Shi et al. demonstrated that FOXF2 down-regulation is common in HCC, and its expression levels are closely correlated with early metastasis and relapse of HCC [14], however, the mechanism has not been described.
In this study, we detected the expression profile of FOXF2 in HCC and focused on the mechanism underlying the correlation between FOXF2 deficiency and metastasis of HCC. A series of in vitro and in vivo experiments were performed in the cells with shRNA-mediated FOXF2 knockdown to elucidate the function of FOXF2 deficiency on HCC metastasis. We found that FOXF2 deficiency induced MET in HCC cells which facilitated the proliferation, colonization of circulating tumor cells and the formation of metastasis.
Materials and methods
Cell culture and reagents
The human hepatic cell line LO2 and human HCC cell line PLC, Huh7 and HepG2 were obtained from Cell Bank of Shanghai Institute of Biological Sciences, Chinese Academy of Sciences. The human HCC cell line MHCC97H was purchased from Shanghai Baili Biotechnology Company. All the cells were maintained in DMEM medium (Gibco, Gaithersburg, MD) supplemented with 10% fetal bovine serum (FBS), and were cultured in 5% CO
Antibodies against E-cadherin, Vimentin and glyce-raldehyde-3-phosphate dehydrogenase (GAPDH) were purchased from Cell Signaling Technology (Beverly, MA). The antibody against FOXF2 was purchased from Santa Cruz.
Animal model
BALB/c-nu mice aged 4 week old were subjected to subcutaneous tumor model and circulating tumor cell colonization model. For subcutaneous tumor model, cells were subcutaneously injected in the flanks of BALB/c-nu mice and observed for tumor growth for a period of 4 weeks. For circulating tumor cell colonization model, cells were injected through the tail vein of nude mice and cultivated for 4 weeks. After euthanasia, the lungs were harvested and the metastatic nodules were counted.
All animal experiments were approved by the Ethics Committee of the First Affiliated Hospital of Chinese PLA General Hospital.
Human HCC tissue specimens
Twenty pairs of HCC tissue and peritumor samples from the Department of Hepatobiliary Surgery, the First Affiliated Hospital of Chinese PLA General Hospital, were included in our research. This study was conducted in accordance with the Declaration of Helsinki (1964) and was approved by the Ethics Committee of the First Affiliated Hospital of Chinese PLA General Hospital. Informed consents were obtained from all participants.
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Total RNA was extracted by using TRIzol (Invitrogen) and was reverse transcripted to cDNA using Primescript RT master mix (TaKaRa, Japan) according to the manufacturer’s instructions. Quantification of target gene expression was performed on ABI PRISM 7300 Sequence Detector by SYBR Green PCR Kit (TaKaRa, Japan). GAPDH was set as an internal control. The primer sequences were: FOXF2 (forward) (5’–AATGCCACTCGCCCTACAC-3’), FOXF2 (reverse) (5’-CGTTCTGGTGCAAGTAGCTCT-3’); GAPDH (forward) (5’-TGTGGGCATCAATGGATTT GG-3’), GAPDH (reverse) (5’-ACACCATGTATTCC GGGTCAAT-3’).
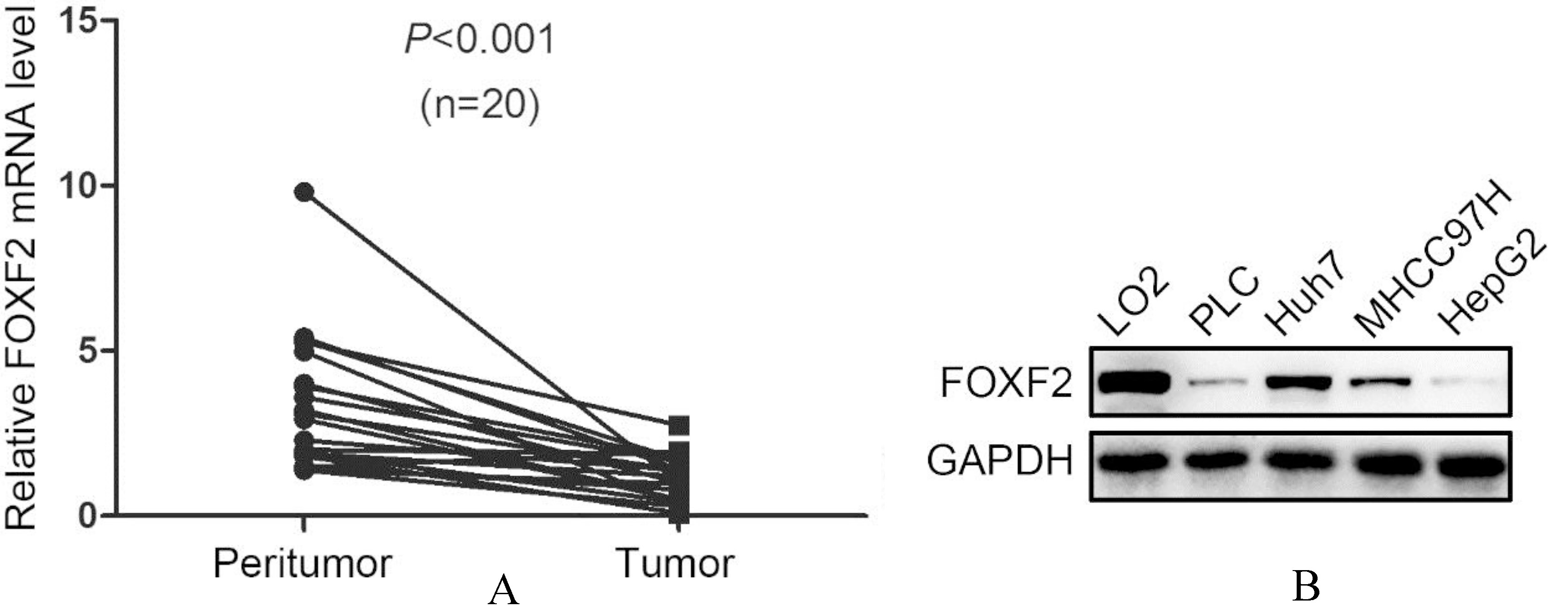
The expression of FOXF2 in HCC tumor tissues and cell lines.
Protein of 10–20
Lentiviral transduction of shRNA
Lentivirus carrying a short hairpin RNA (shRNA) targeting FOXF2 was synthesized by Shanghai Gene-chem Company Ltd (GeneChem, Shanghai, China). FOXF2 targeting sequence was GTCCTCAACTTCA ATGGGATT. A shRNA nontargeting human gene was used as control. The lentiviral transduction was conducted following the manufacturer’s instructions to generate Huh7 cells with stable FOXF2 knock-down.
Wound healing assay
Wound healing assay was conducted by using Wou-nd Healing culture inserts (Ibidi, Munich, Germany) following the manufacturer’s instructions. Wound closure rate was calculated to represent the cell migration ability.
Transwell assay
Cell invasion ability in vitro was assessed by using Matrigel-coated transwell inserts (BD Biosciences) following the manufacturer’s instructions. Cells invaded to the opposite side the membranes were coun-ted.
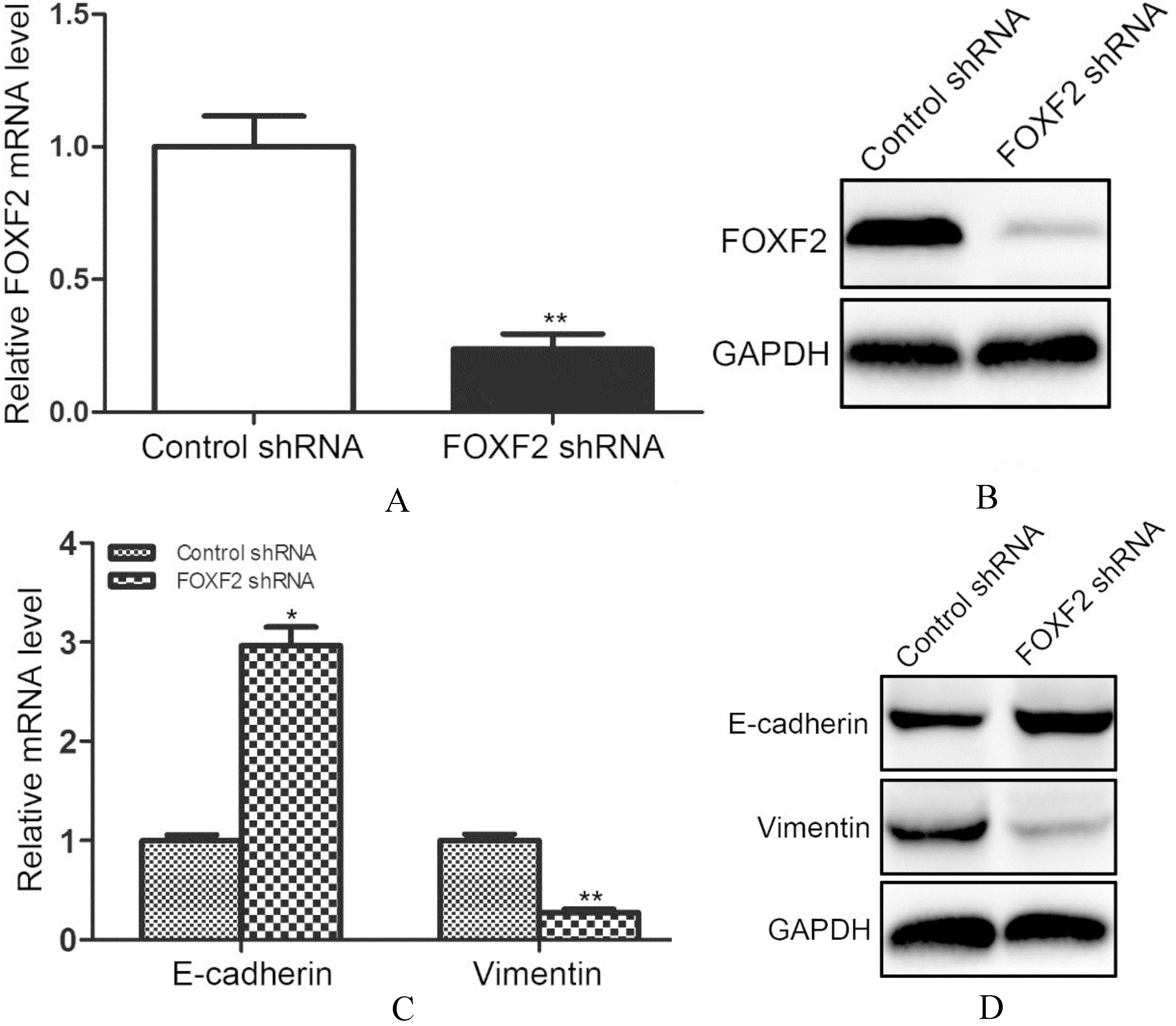
FOXF2 deficiency increased E-cadherin and decreased Vimentin.
Cell proliferation ability was assessed by colony formation assay and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. For colony formation assay, 1
Hematoxylin-eosin (H&E) staining
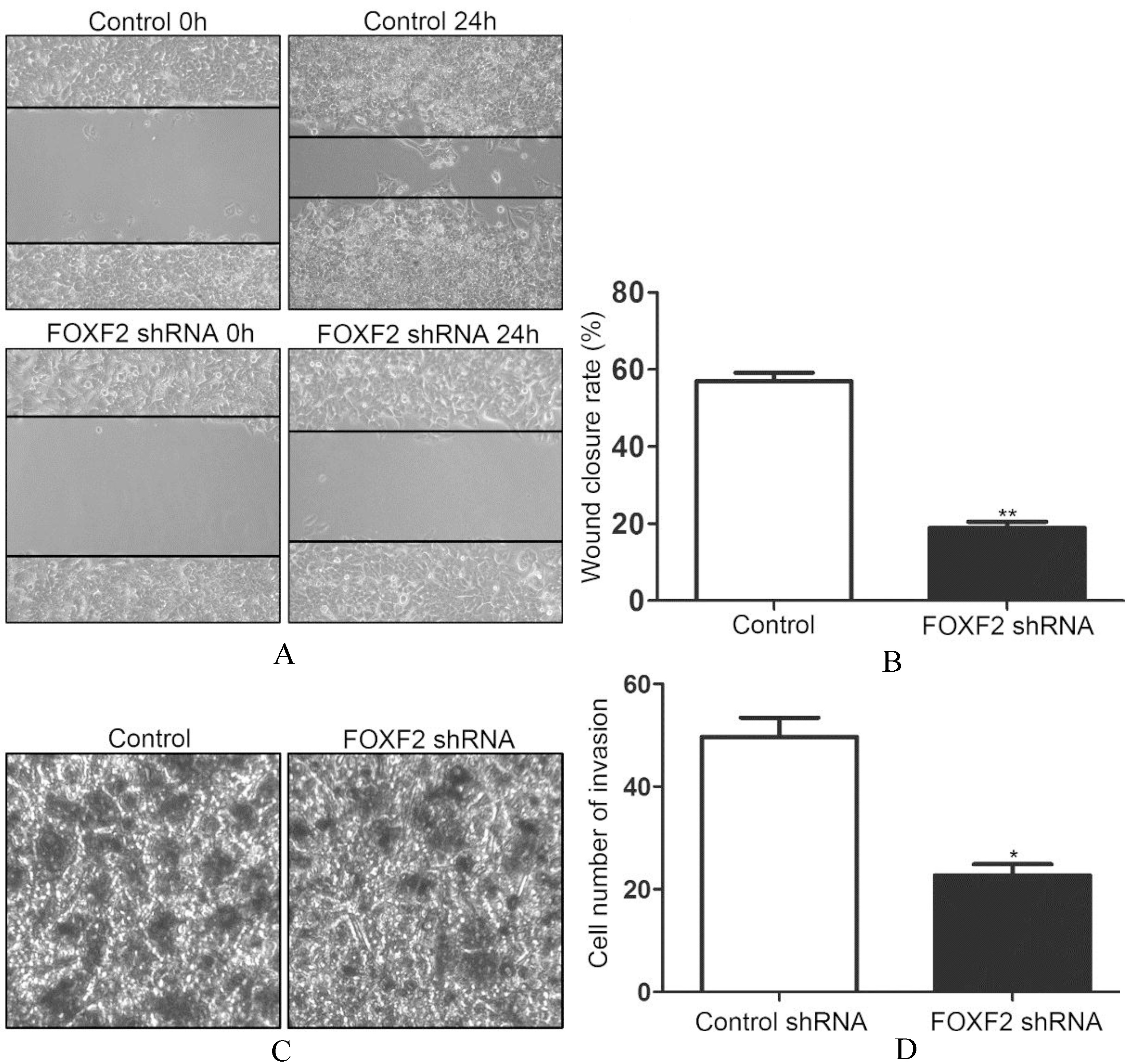
FOXF2 deficiency inhibited migration and invasion of Huh7 cells.
The nude mouse lungs were fixed in 4% paraformal-dehyde and embedded in paraffin. The slices of 5-
The data were expressed as mean
Results
FOXF2 was down-regulated in HCC tumor tissues and cell lines
To reveal the role of FOXF2 in HCC, we first detected the expression profile of FOXF2 in 20 pairs of HCC tissues and peritumor tissues by qRT-PCR. The results showed that FOXF2 was down-regulated in HCC tumor tissues (Fig. 1A). We also screened the FOXF2 expression in 4 different HCC cell lines and a normal hepatic cell line LO2, and observed a notable decrease of FOXF2 in HCC cells (Fig. 1B). It was noticed that HCC cells with a mesenchymal phenotype (Huh7 and MHCC97H) tended to have more FOXF2 than epithelial HCC cells (PLC and HepG2).
The results in HCC were consistent with the findings by Shi et al. that FOXF2 was down-regulated in HCC tissues and cell lines.
FOXF2 deficiency increased E-cadherin and decreased Vimentin.
To further explore the effect of FOXF2 deficiency on HCC progression. We used Huh7 cells for further experiments and transfected Huh7 cells with lentivirus carrying FOXF2 shRNA sequences. The knock-down efficiency was verified by qRT-PCR and western blotting, respectively. The qRT-PCR results showed that the interfering rate was around 70% (Fig. 2A). The western blotting results confirmed decreased FOXF2 in Huh7 cells transfected with FOXF2 shRNA (Fig. 2B).
The research by Shi et al. demonstrated that FOXF2 deficiency was correlated with shorter disease-free survival rate in HCC patients. The down-regulated FOXF2 was predictive of recurrence and metastasis of HCC. Because FOXF2 was involved in epithelial-mesenchymal interaction which was a significant me-chanism underlying tumor metastasis, our researches focused on the changes of cell phenotypes. We first detected the two regular cell phenotype markers E-cadherin and Vimentin in Huh7 cells with FOXF2 shRNA transfection. We found increased E-cadherin and decreased Vimentin mRNA levels after inhibiting FOXF2 (Fig. 2C). The protein changes were consistent with that in mRNA levels (Fig. 2D).
The above results indicated that the down-regulation of FOXF2 might induce a mesenchymal to epithelial phenotypic change in HCC cells
FOXF2 deficiency inhibited migration and invasion of Huh7 cells
Accompanied by the phenotypic changes, the migration and invasion capacity of tumor cells also changes. So in our further researches, we accessed the migration and invasion ability of Huh7 cells after inhibiting FOXF2. By wound healing assay, we found that FOXF2 deficiency impeded the migration of Huh7 cell (Fig. 3A). The decreased wound closure rate was significantly different (Fig. 3B). We also accessed the invasion capacity of Huh7 with FOXF2 deficiency by transwell assay. The results showed decreased invasion of Huh7 cells after inhibiting FOXF2 (Fig. 3C). The changes was also significantly different (Fig. 3D).
Based on the above results, we speculated that FOXF2 deficiency induced MET in HCC cells.
FOXF2 deficiency promoted Huh7 cell proliferation and the growth of subcutaneous tumor in nude mice
When tumor cells underwent MET, they usually acquired enhanced proliferation to form metastasis, so we further evaluated the proliferative capacity of Huh7 cells transfected with FOXF2 shRNA. We found increased colony formation of Huh7 cells with stable FOXF2 knock-down (Figs 4A and B). The cell growth curves demonstrated that FOXF2 knock-down significantly promoted the proliferation of Huh7 cells (Fig. 4C). To test whether FOXF2 deficiency could alter the proliferation of Huh7 cells in vivo, Huh7 cells with stable FOXF2 knock-down were injected subcutaneously in the flank of nude mice. Tumor volumes were measured 4 weeks after injection which showed increased volume in the group of Huh7 cells with FOXF2 knock-down (Fig. 4D).
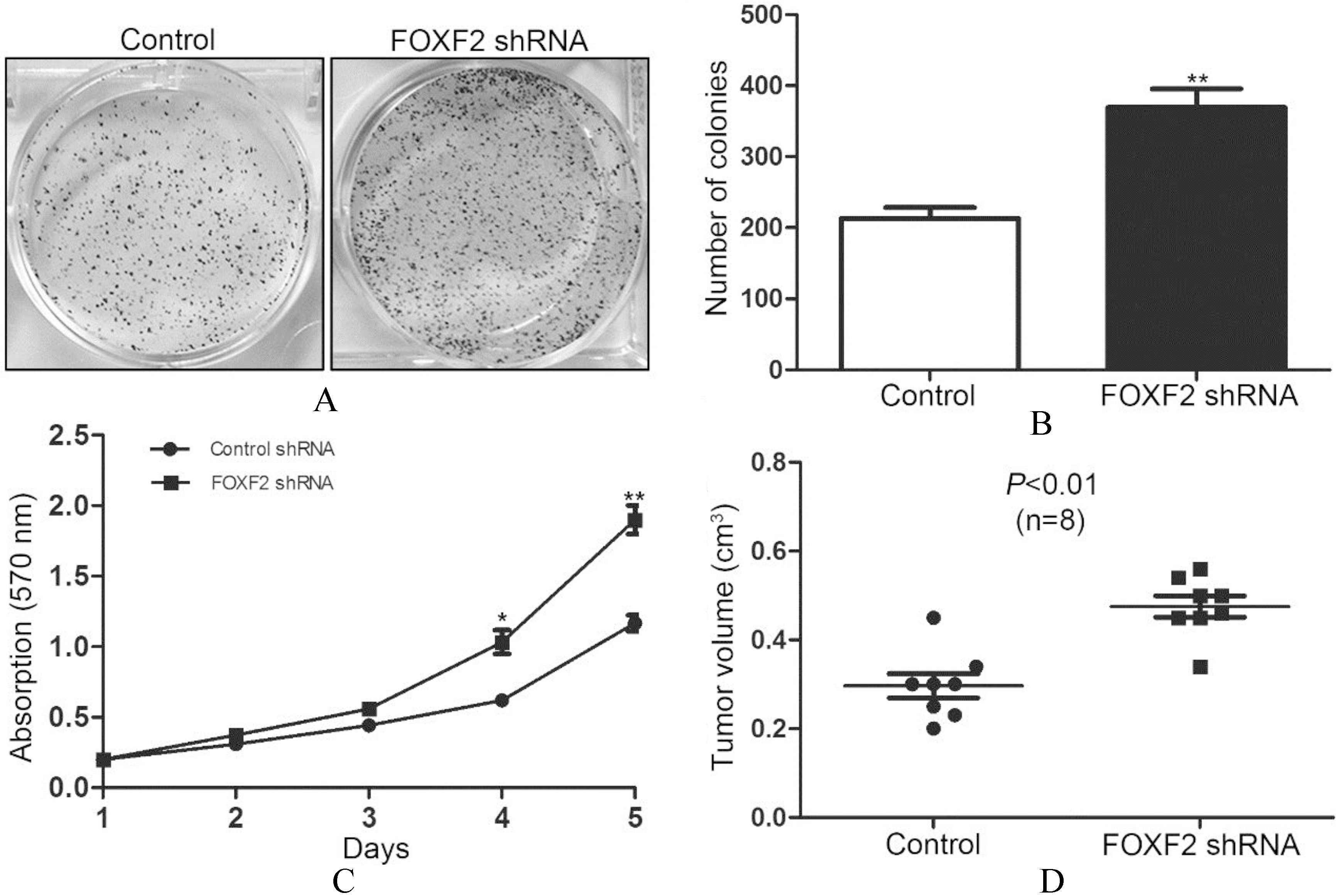
FOXF2 deficiency promoted Huh7 cell proliferation and the growth of subcutaneous tumor in nude mice.
The research by Ocana et al. demonstrated that MET process was necessary for circulating tumor cells to colonize and form metastasis [15]. Tumor cells that underwent EMT had to go through a reversed transversion to acquire an epithelial phenotype again to form metastatic nodules, so we tested whether FOXF2 deficiency affect the colonization of circulating HCC cells. Huh7 cells with stable FOXF2 knock-down and control cells were injected through the tail vein of nude mice. Four weeks after injection, the metastatic nodules in the lung were counted. We found that FOXF2 deficiency increased the formation of lung metastatic nodules (Fig. 5A), which was confirmed by H&E staining of the lung sections (Fig. 5B).
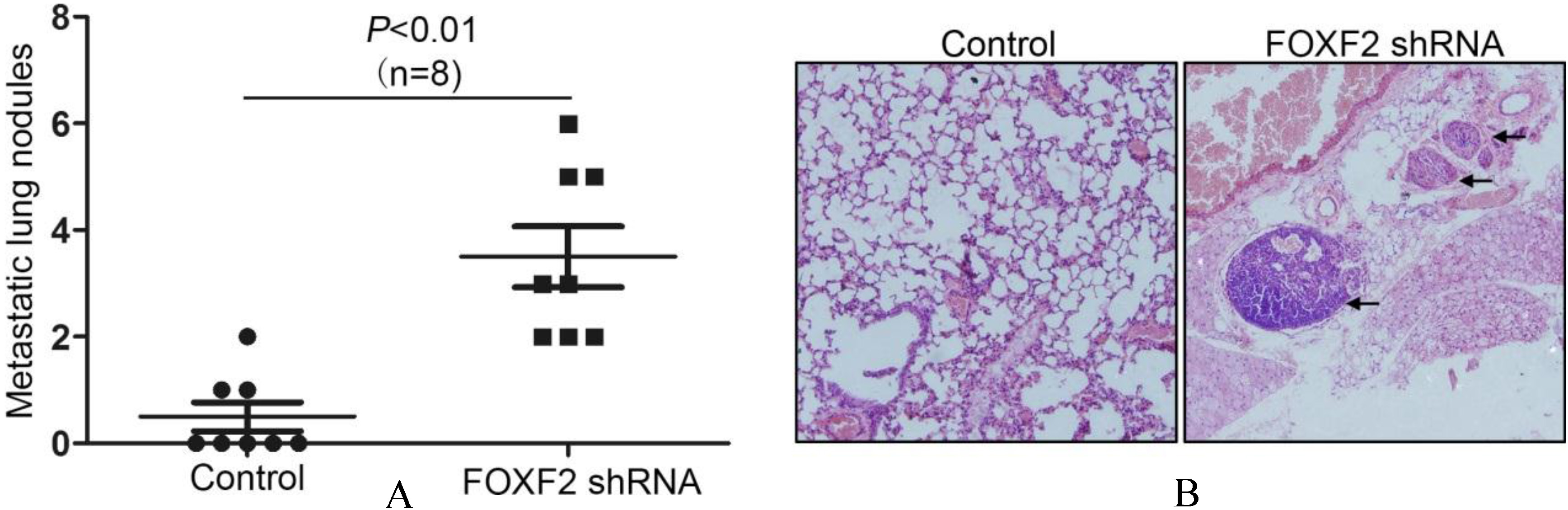
FOXF2 deficiency promoted the colonization of circulating Huh7 cells and the formation of metastatic nodules in nude mouse lung.
In our research, we found that FOXF2 was down-regulated in HCC tissues and cell lines, and this was consistent with existing studies [14]. Then we focused on the possible mechanisms underlying the correlation between FOXF2 deficiency and the early metastasis by a series of in vitro and in vivo assays. We found that the FOXF2 deficiency in HCC cells increased E-cadherin and reduced Vimentin, which indicated a mesenchymal to epithelial phenotypic change. The down-regulation of FOXF2 impeded HCC cell migration and invasion capacity, but promoted the proliferation of HCC cells and the growth of subcutaneous tumors in nude mice. FOXF2 deficiency enhanced the colonization of circulating HCC cell, thus promoted the formation of metastatic nodules.
The transcription factor FOXF2, a member of FOX superfamily, is involved in tissue development, ECM synthesis and epithelial-mesenchymal interactions.FOXF2 has been revealed as a tumor suppressor and the down-regulation of FOXF2 correlated with poor prognosis in divergent cancer types [9, 14]. The role of FOXF2 in regulating the epithelial-mesenchymal interaction, migration and invasion was controversial. In cancer metastasis, many researches showed that decreased FOXF2 promoted migration and invasion of tumor cell by inducing EMT [13, 16, 17, 18]. However, the recent research by Kundu et al. demonstrated that inhibited FOXF2 impeded cell invasion of lung cancer. FOXF2 was increased in mesenchymal-like metastatic lung cancer cells. The FOXF2 expression could induce EMT, migration, invasion in lung cancer cells [19]. The research by Lo et al. also showed that FOXF2 was required for cell migration and invasion of basal-like breast cancer cells by regulating the expression of genes implicated in EMT regulation [20]. Our research showed that decreased FOXF2 inhibited the mesenchymal features of HCC cell such as the expression of Vimentin and the migration and invasion capacity. This was consistent with the study by Kundu and Lo.
In our research, we also found that FOXF2 deficiency promoted cell proliferation of HCC cell. The enhanced cell viability might be the results of Wnt/
So based on the results that FOXF2 deficiency inhibited cell invasiveness and promoted cell proliferation of HCC cells, we speculated that FOXF2 deficiency induced MET in HCC cells. MET was recognized as a revere process of EMT, in which mesenchymal tumor cells disseminating from the origin site acquired the epithelial phenotypes again to form metastatic neoplasm [23]. The EMT progression facilitated tumor cells with increased motility and invasiveness, promoted the dissemination from original tumor sites and infiltration into circulation [24]. However, those circulating tumor cells could not form metastasis without the happening of the reverse process MET. Tumor cells underwent MET to re-differentiate and to acquire enhanced proliferation during colonization and formation of metastasis [25]. So MET was necessary for efficient colonization and macrometastasis [15, 26].
Several studies have been conducted to elucidate how FOXF2 was down-regulated in tumor cells. The decreased FOXF2 level was often the results of some increased microRNAs. The research by Kundu et al. demonstrated that the miR-183/96/182 cluster could directly target FOXF2 to inhibit the migration and invasion of lung cancer cells [19]. In colorectal cancer, miR-182 could bind to the 3’ untranslated region (3’UTR) of FOXF2 mRNA to promote cell growth and invasion [18]. MiR-182-5p was also reported to be able to inhibit FOXF2 in prostate cancer [17]. MiR-301 was another miRNA targeting FOXF2 in breast cancer [27]. In HCC, miRNA-519a was found to be up-regulated. MiRNA-519a enhanced the proliferation and inhibited apoptosis of HCC cells by targeting FOXF2 [28]. The way how FOXF2 is down-regulated in HCC has not been extensively described yet. Besides miRNA-519a, other three miRNAs mentioned above are also promising ones that could target FOXF2 in HCC. MiR-183/96/182, miR-182 and miR-301 were reported to be up-regulated in HCC and correlate with poor prognosis of HCC. Those miRNAs have a vital role in modulating the cell growth and invasion of HCC cells [29, 30, 31]. Leung et al. found that miR-183/96/182 was over-expressed in HCC tissues and correlated with metastatic features including presence of microvascular invasion, advanced tumor differentiation, and shorter recurrence-free survival of HCC. What was more, the downstream target of miR-183/96/182 was FOXO1, another FOX family member [32]. We speculate that the down-regulated FOXF2 in HCC is also the consequence of some aberrantly expressed miRNAs, including miR-519a and miR-183/96/182.
In summary, we conclude that decreased FOXF2 is common in HCC tissues and cell lines. The FOXF2 deficiency in cells induces MET thus promotes the metastasis of HCC. However, the way how FOXF2 is down-regulated in HCC needs further investigations.