
Abstract
Nitric oxide/cyclic guanosine monophosphate (cGMP) signaling is compromised in Alzheimer’s disease (AD), and phosphodiesterase 5 (PDE5), which degrades cGMP, is upregulated. Sildenafil inhibits PDE5 and increases cGMP levels. Integrating previous findings, we determine that most doses of sildenafil (especially low doses) likely activate peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α) via protein kinase G-mediated cyclic adenosine monophosphate (cAMP) response element binding protein (CREB) phosphorylation and/or Sirtuin-1 activation and PGC1α deacetylation. Via PGC1α signaling, low-dose sildenafil likely suppresses β-secretase 1 expression and amyloid-β (Aβ) generation, upregulates antioxidant enzymes, and induces mitochondrial biogenesis. Plus, sildenafil should increase brain perfusion, insulin sensitivity, long-term potentiation, and neurogenesis while suppressing neural apoptosis and inflammation. A systematic review of sildenafil in AD was undertaken. In vitro, sildenafil protected neural mitochondria from Aβ and advanced glycation end products. In transgenic AD mice, sildenafil was found to rescue deficits in CREB phosphorylation and memory, upregulate brain-derived neurotrophic factor, reduce reactive astrocytes and microglia, decrease interleukin-1β, interleukin-6, and tumor necrosis factor-α, decrease neural apoptosis, increase neurogenesis, and reduce tau hyperphosphorylation. All studies that tested Aβ levels reported significant improvements except the two that used the highest dosage, consistent with the dose-limiting effect of cGMP-induced phosphodiesterase 2 (PDE2) activation and cAMP depletion on PGC1α signaling. In AD patients, a single dose of sildenafil decreased spontaneous neural activity, increased cerebral blood flow, and increased the cerebral metabolic rate of oxygen. A randomized control trial of sildenafil (ideally with a PDE2 inhibitor) in AD patients is warranted.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is the leading cause of dementia worldwide, and AD patients and their families urgently require novel therapeutics to prevent and slow the progression of this devastating disorder. Hallmarks of AD include amyloid-β (Aβ) peptide secretion and deposition into neuritic plaques, tau protein hyperphosphorylation and neurofibrillary tangle formation, metal ion dyshomeostasis [1–9], oxidative stress and lipid, nucleic acid, and protein damage [10–13], abortive cell cycle reentry [14–26], neuroinflammation and microbial dysbiosis [27–33], insulin resistance [34, 35], cerebrovascular dysfunction [36–38], synaptic dysfunction [39, 40], neuronal loss, endoplasmic reticulum stress [41–44], and mitochondrial dysfunction [45–48].
Sildenafil (Viagra) is a drug used to treat erectile dysfunction and pulmonary arterial hypertension that inhibits phosphodiesterase 5 (PDE5) (Fig. 1). PDE5 degrades cyclic guanosine monophosphate (cGMP). Upstream of cGMP, normally, the amino acid L-arginine is converted by three varieties of the enzyme nitric oxide synthase (NOS) into nitric oxide (NO). NO is a small cell-permeable gas molecule that diffuses across the plasma membrane and activates soluble guanylyl cyclase (sGC). sGC converts guanosine triphosphate (GTP) into cGMP [49]. However, in AD, the activity of the NOS/NO/cGMP pathway is severely impaired. NOS activity is significantly decreased in AD patients’ superior frontal gyri and hippocampi compared to age-matched controls [50], even though aberrant neuronal NOS (nNOS) protein expression has been observed in a subpopulation of isocortical pyramidal neurons in AD patients’ brains [51, 52] and the intensity of astrocyte endothelial NOS (eNOS) and inducible NOS (iNOS) expression had increased in AD patients’ deep cortical layers [52, 53]. NO-induced soluble sGC (but not basal sGC or particulate guanylyl cyclase) activity was found to be decreased by 50% in AD patients’ superior temporal cortices compared to controls [54]. cGMP levels were found to be significantly lower in AD patients’ cerebrospinal fluid (CSF) compared to controls, and decreases in levels of cGMP correlated with CSF Aβ42 levels [55], comorbid depression [56], and cognitive decline as measured by Mini-Mental State Examination (MMSE) [55, 57].
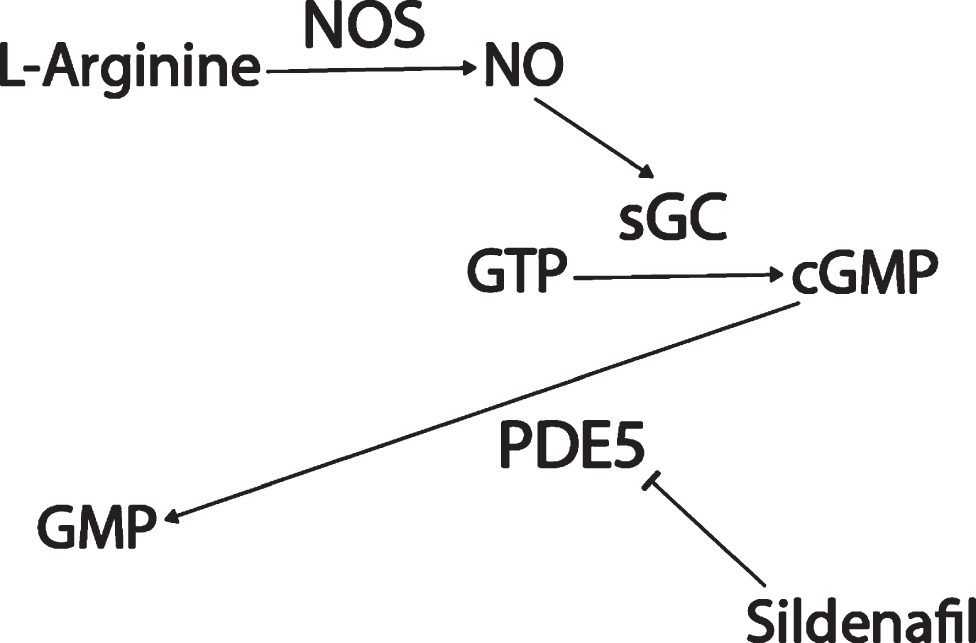
Sildenafil mechanism of action.
Potentially contributing to this cGMP depletion, PDE5 protein levels have been found to be significantly upregulated in AD patients’ temporal cortices [55], and PDE5 mRNA levels are significantly elevated in AD patients’ entorhinal cortices compared to controls according to a meta-analysis of mRNA datasets (p = 0.001, FDR = 0.018; Fig. 2) [58]. Granted, low PDE5 mRNA expression in the human CNS has been reported relative to peripheral tissue [59–61], and no specific hybridization signal was observed for PDE5 mRNA in the brains of aged and AD patients using radioactive in situ hybridization histochemistry in one study, drawing skepticism as to whether PDE5 inhibition has relevance to AD [62–64]. Regardless though, the weight of the evidence suggests that PDE5 is expressed (albeit relatively less than in peripheral tissues) in the normal human brain [58–61] and that, moreover, it is upregulated in the AD entorhinal and temporal cortices [55, 58], supporting the notion that PDE5 may be an important therapeutic target in AD.
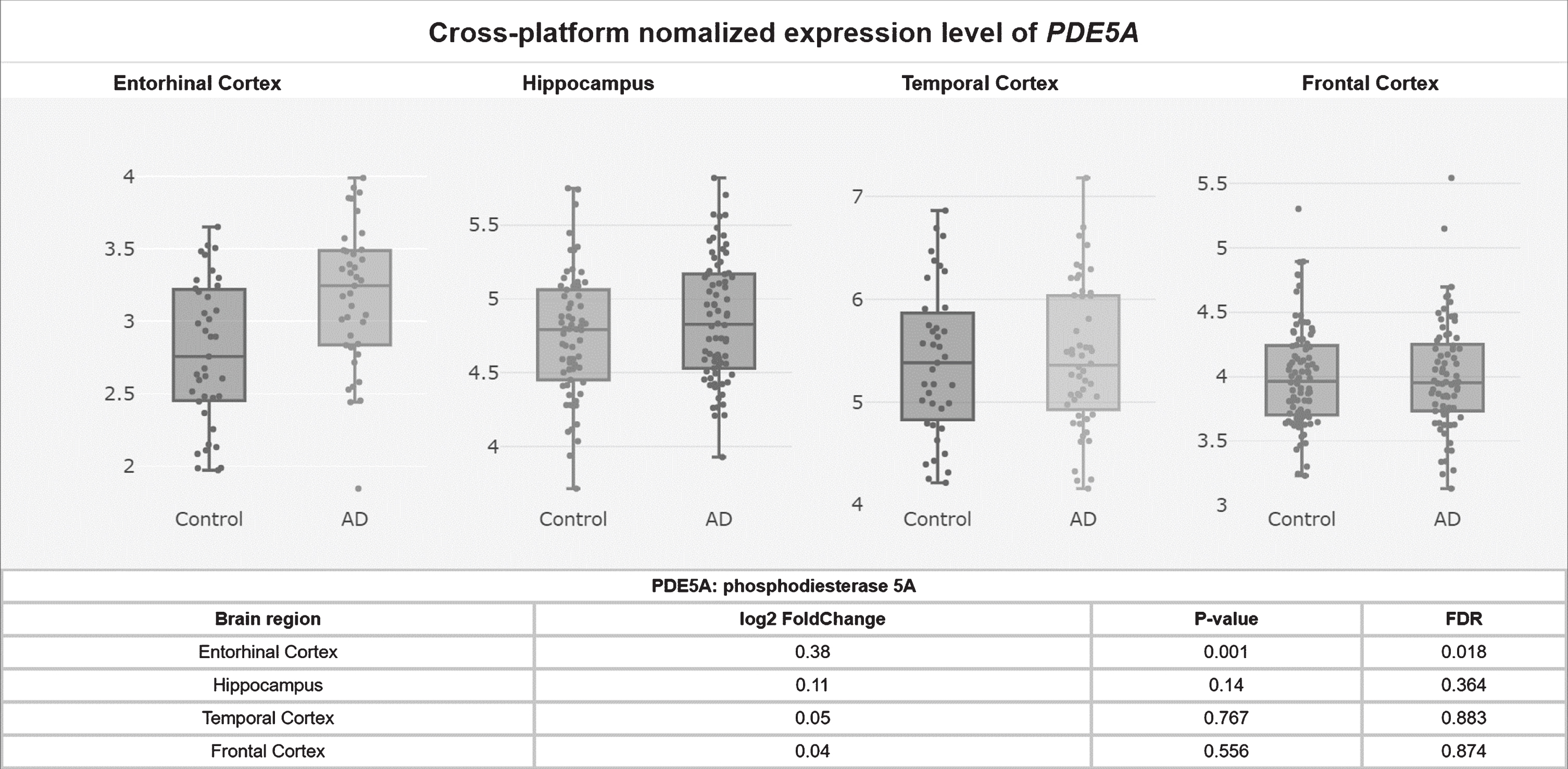
PDE5A mRNA levels upregulated in the AD entorhinal cortex.
cGMP/PGC1α signaling
This multi-mechanism NOS/NO/cGMP signaling dysfunction is an important therapeutic target in AD for multiple reasons. One reason is that cGMP is responsible for increasing the expression and activity of peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α). PGC1α overexpression (or low-dose NO) suppresses the expression of β-secretase 1 (BACE1) [65], the rate-limiting enzyme in Aβ generation, suggesting that PGC1α activity suppresses Aβ generation. In addition, PGC1α is a master transcriptional regulator of mitochondrial biogenesis, oxidative respiration [66, 67], fatty acid β-oxidation [68], and antioxidant defense [69]. PGC1α upregulates multiple antioxidant genes, including mitochondrial manganese superoxide dismutase (MnSOD), when bound to Forkhead box O3a (Foxo3a) and deacetylated by Sirtuin-1 (SIRT1) [69]. However, PGC1α protein levels are significantly lower in AD patients’ hippocampi compared to controls [48], and mitochondrial biogenesis and MnSOD expression are impaired there [45, 48]. Sildenafil has the potential to reverse the hippocampal PGC1α suppression in AD. 10μM sildenafil treatment for 24 hours in vitro and 0.3 mg/kg sildenafil in vivo have been shown to induce PGC1α expression and mitochondrial biogenesis by increasing cGMP in renal proximal tubular cells and mouse renal cortex, respectively [70]. In addition, sildenafil has been shown to upregulate SOD and catalase activities in rat liver (1.48 mg/kg sildenafil) and human blood (100 mg sildenafil one time dosage) [71, 72].
Multiphasic regulation of PGC1α by sildenafil and cGMP
NOS/NO/sGC/cGMP signaling upregulates PGC1α in diverse cell types, including not only renal proximal tubular cells [70], but also brown adipocytes, U937 monocytic cells, HeLa cervical cancer-derived cells, white 3T3-L1 adipocytes [73, 74], and probably neurons [75]. In mice, subcortical brain tissue responded to hypoxia with PGC1α upregulation (as well as with mitochondrial biogenesis) in a manner that depended on nNOS expression [75], so the nNOS/NO/cGMP/PGC1α pathway does appear to be active in neurons.
An important caveat to the claim that sildenafil and cGMP activate PGC1α though is that NOS/NO/cGMP appears to be a multiphasic rheostat of PGC1α signaling whose outcome depends on cGMP concentration, duration, and crosstalk with cAMP signaling mediated by phosphodiesterase 3 (PDE3) and phosphodiesterase 2 (PDE2) activity. Low concentrations of cGMP compete for the active site of PDE3 [76], thereby impeding PDE3 from degrading cAMP and increasing cAMP levels. By contrast, high cGMP concentrations allosterically activate PDE2, thereby promoting cAMP degradation [76, 77]. Mirroring this, low-dose sildenafil (up to 1μM) inhibits PDE3, resulting in cAMP accumulation [76], whereas high-dose sildenafil (10 and 100μM) activates PDE2, resulting in cAMP degradation [76] and suppression of the cAMP/EPAC/adenosine monophosphate-activated protein kinase (AMPK) pathway [76, 79]. For this reason, excessively high doses of sildenafil resulted in inferior therapeutic responses compared to low doses in preclinical models of non-alcoholic hepatic steatosis and obesity [76]. This caveat may be relevant to AD as well. The dose-dependent effect of sildenafil and cGMP signaling on cAMP levels should affect PGC1α regulation since cAMP is required for robust PGC1α expression and mitochondrial biogenesis in AD [48, 67]. On top of this, although NO/sGC/cGMP signaling typically promotes PGC1α activity, it also simultaneously activates the protein kinase G (PKG)/phosphoinositide 3-kinase (PI3K)/Akt signaling cascade, which decreases PGC1α activity [80–83]; opposing this negative regulation of PGC1α by cGMP, cAMP inhibits Akt via PKA and Rap1b (Fig. 4) [84]. In other words, cGMP per se simultaneously increases and decreases PGC1α signaling by distinct mechanisms, with crosstalk with cAMP pathways probably determining whether PGC1α is activated or inhibited overall. Therefore, since high doses of sildenafil activate PDE2 and decrease cAMP levels, high-dose sildenafil would be expected to be less effective than low-dose sildenafil at activating PGC1α, inducing mitochondrial biogenesis, upregulating antioxidant genes, and downregulating BACE1 [76].
Low and high but not moderate cGMP levels activate PGC1α?
Consistent with the dose-dependent effect of sildenafil and cGMP on PGC1α, 10μM of 8-Br-cGMP treatment for 24 hours upregulated PGC1α mRNA expression and mitochondrial biogenesis in renal proximal tubular cells [70]. By contrast, in endothelial cells, 100μM 8-Br-cGMP treatment for 6–24 hours downregulated PGC1α [80, 83]. In addition, treatment for less than 12 hours with NO donors decreased PGC1α expression, whereas treatment for 24 hours or more increased PGC1α expression [83]. NO-triggered PGC1α downregulation and consequent reactive oxygen species (ROS) production were required for endothelial cell migration, an effect that was mediated by NO-activated PI3K/Akt signaling and Akt phosphor-inhibition of Foxo3a [80].
Surprisingly though, much higher dosages of cGMP than 100μM upregulate PGC1α like lower doses do: for example, PGC1α was upregulated in brown adipocytes treated with 1 mM 8-Br-cGMP for 4 days [73]. In human monocytic U937 cells, rat L6 myotubes, and rat PC12 neurosecretory cells, 3 mM 8-Br-cGMP treatment every day for 6 days upregulated PGC1α, as well as Nrf1 and Tfam, mitochondrial proteins Cox IV and Cytochrome C, and mitochondrial DNA content [66]. Furthermore, 6 days of 3 mM 8-Br-cGMP in these three cell types resulted in increased oxidative phosphorylation-coupled oxygen consumption [66].
Therefore, there appears to be a U-shaped dose-response curve between cGMP concentrations and PGC1α expression, with PGC1α being downregulated by 100μM cGMP for 6–24 hours [80, 83] but upregulated by 10μM cGMP or 1–3 mM cGMP for 24 hours or longer (Fig. 3) [66, 73]. This might be because 100μM cGMP corresponds to the 10–100μM dosage of sildenafil that is sufficient to activate PDE2 and lower cAMP [76], whereas much higher dosages of cGMP and sildenafil nevertheless stimulate mitochondrial biogenesis and PGC1α transcription [66, 73] because supraphysiological cGMP signaling is sufficient to overcome the cAMP depletion to induce robust PGC1α activity independently of cAMP, perhaps via cAMP response element binding protein (CREB) phosphorylation and SIRT1 activation (see next subsection). If so, this would not be unprecedented: in fibroblasts, PDE2 overexpression was sufficient to lower cAMP levels and induce a transition into a pro-fibrotic myofibroblast phenotype, an alternation that cGMP elevating agents reversed, suggesting that sufficiently high cGMP levels can overcome the deficits to cellular signaling induced by PDE2-mediated cAMP depletion [77]. Therefore, it appears that, for optimal PGC1α expression, mitochondrial biogenesis, and BACE1 downregulation, either sufficiently low or high sildenafil dosages must be used, or sildenafil must be co-administered with a PDE2 inhibitor.
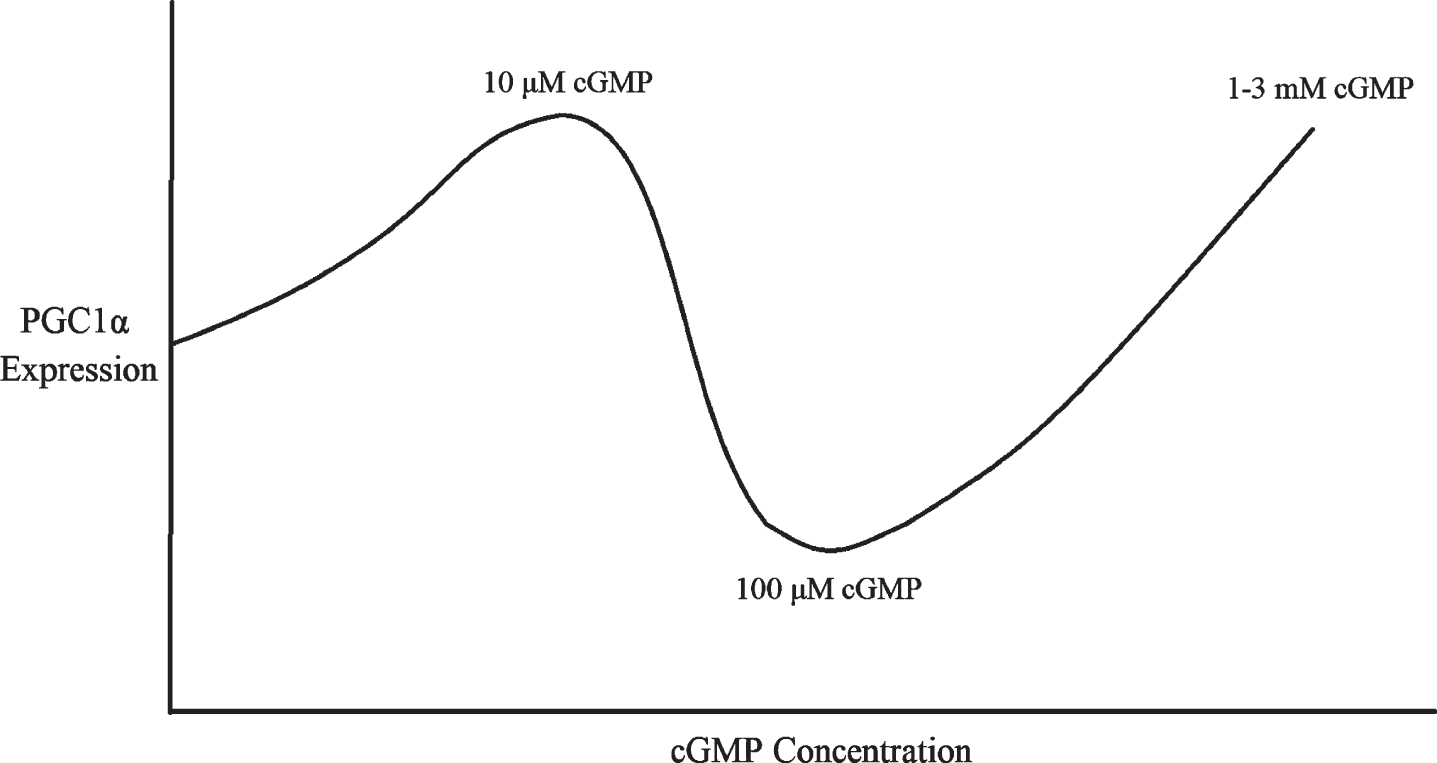
Multiphasic dose response of PGC1α expression to cGMP concentration.
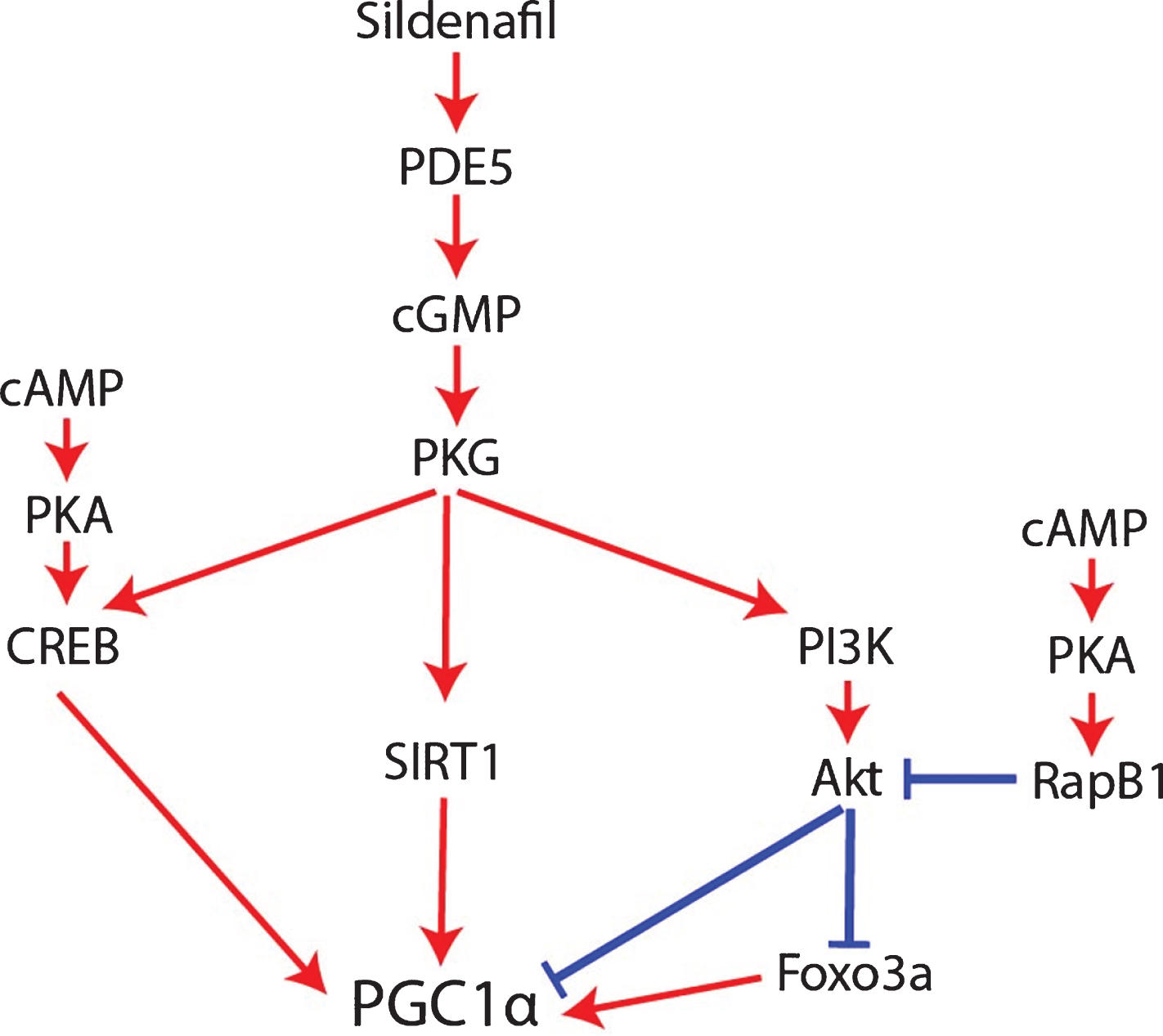
How sildenafil and cGMP may regulate PGC1α.
In addition to this complex but clinically relevant dose-specific effect, the mechanism by which NO/cGMP typically upregulates PGC1α remains unresolved [67]. Two potential mechanisms involving either CREB or SIRT1 will now be described.
How sildenafil and cGMP activate PGC1α
In the hippocampus during long-term potentiation and in other neural tissues, both the cGMP/PKG and the cAMP/PKA pathways contribute to CREB phosphorylation [85–90], and PKA-mediated CREB phosphorylation promotes PGC1α transcription [67], so cGMP/PKG/CREB signaling might promote PGC1α expression as well. In addition, cGMP may contribute to the post-translational regulation of PGC1α. Once transcribed and translated, the stability, subcellular localization, and co-activator activity of PGC1α proteins are regulated by multiple post-translational modifications. For example, PGC1α can be inhibited via phosphorylation by Akt [81, 82], S6 Kinase [91], or glycogen synthase kinase 3β (GSK3β) [74, 92], or via acetylation by general control of amino acid synthesis 5 (GCN5) [81, 93–95] or p300 [96]. Conversely, PGC1α can be activated via phosphorylation by adenosine monophosphate activated protein kinase (AMPK) [97–101] or p38 mitogen activated protein kinase (MAPK) [101, 102], via methylation by protein arginine methyltransferase 1 (PRMT1) [103], or via deacetylation by SIRT1 (Fig. 5) [69, 104–107]. Interestingly given the SIRT1-mediated activation of PGC1α, sildenafil has been shown to upregulate SIRT1 in heart, cardiomyocytes [108], serum, and subcutaneous adipose tissue [109]. cGMP analogues upregulated SIRT1 expression in white adipose tissue [110, 111] and in osteoblasts [112]. Mice with osteoblast-specific PKGII overexpression exhibited increased SIRT1 expression [112]. The PKG inhibitor KT-5823 blocked the relief of spinal allodynia induced by resveratrol, a SIRT1 activator [113, 114]. In hypoxic myocardial cells, 1μM sildenafil treatment decreased PGC1α protein acetylation [115, 116]. Therefore, it is possible that sildenafil may promote PGC1α deacetylation in some contexts via cGMP/PKG/SIRT1 signaling (Fig. 5).
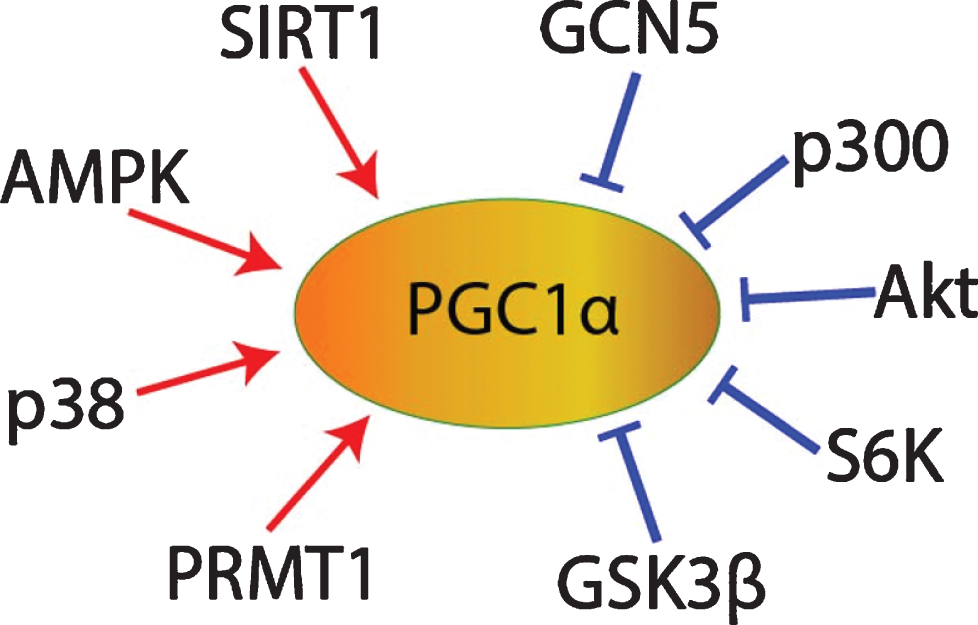
PGC1α post-translational regulation.
Benefits of sildenafil in AD
Since most doses of sildenafil/cGMP activate PGC1α and PGC1α signaling induces mitochondrial biogenesis [67, 70], upregulates antioxidant enzymes [69], and suppresses BACE1 expression [65], sildenafil should provide significant benefits to patients with AD. In addition to its PGC1α-specific benefits, sildenafil promotes smooth muscle relaxation and vasodilation via cGMP [117], which might provide additional benefit to patients with AD since hypoperfusion is also a significant impairment in AD patients’ brains [37, 118]. Sildenafil suppresses apoptosis in hypoxic neurons [117, 119] and promotes neurogenesis [120–123], so it might slow the loss of AD neurons and promote the replenishment of new ones. Furthermore, sildenafil improves insulin sensitivity and endothelial inflammation in patients [124–126], so sildenafil might also promote insulin sensitivity and suppress inflammation in AD brains. Since cGMP/PKG signaling mediates long-term potentiation via CREB phosphorylation [85–90, 117], sildenafil should improve the learning and memory impairments associated with AD. Therefore, in theory, most doses of sildenafil should improve multiple hallmarks of AD, including excessive Aβ generation, impaired mitochondrial biogenesis, oxidative stress, neuroinflammation, hypoperfusion, insulin resistance, neuron loss, insufficient neurogenesis, and memory deficits.
Sildenafil and AD comorbidities and risk factors
It is also important to consider the effects of sildenafil on common AD comorbidities and/or risk factor conditions, such as type II diabetes, cardiovascular diseases, and depression, since many AD patients suffer from one or more of these conditions.
Regarding the effect of sildenafil in depression, NOS/NO/sGC/cGMP and serotonin signaling tend to oppose each other. cGMP triggers cerebral vasodilation [127], whereas serotonin induces cerebral vasoconstriction [128, 129]. NOS/NO/sGC/cGMP/PKG signaling activates the serotonin transporter (SERT), inducing serotonin reuptake [130–132]. For this reason, sildenafil might be expected to make selective serotonin reuptake inhibitor (SSRI) antidepressants less effective. However, there do not appear to be any reports of this being the case, and sildenafil has been used safely and successfully to treat erective dysfunction in patients taking SSRIs [133]. Moreover, sildenafil itself has been shown to exert an antidepressant effect in mice [134].
Regarding the effects of sildenafil in type II diabetes, it has been found in a randomized, double-blind, placebo-controlled study that 25 mg thrice daily for 3 months sildenafil increases insulin sensitivity in patients with pre-diabetes, indicating that sildenafil might be beneficial for patients with AD and type II diabetes [125]. Regarding the effects of sildenafil and cardiovascular diseases, despite early concerns [135], sildenafil usage does not appear to contribute to myocardial infarction or sudden cardiac death risk [136]. In fact, treatment of erectile dysfunction in patients who had had a myocardial infarction with PDE5 inhibitors (but not alprostadil) correlated with a reduced risk of mortality and hospitalization for heart failure (n = 43,145) [137]. Preclinically, in a mouse model of hypercholesterolemia, sildenafil decreased aortic atherosclerotic plaques by 40% [138]. Furthermore, sildenafil decreases cardiac hypertrophy [139]. Therefore, sildenafil treatment in AD patients with comorbid cardiovascular diseases would be expected to be safe and potentially beneficial.
MATERIALS AND METHODS
To determine the current progress in studying sildenafil and AD, we searched PubMed for “sildenafil Alzheimer’s.” Both preclinical and clinical studies were reviewed. Results that were not about the effect of sildenafil on patients or preclinical models with AD (e.g., studies about the interaction between cGMP and Aβ in long-term potentiation) were discarded. To ensure clinical relevancy, studies about derivates of sildenafil were also discarded.
RESULTS
As per the methods section, two in vitro studies, ten rodent studies, one systematic review, and two pilot studies in patients were included. Overall, all the studies supported the use of sildenafil in AD (see Table 1 for a summary of results).
Literature review results
HT-22 hippocampal neuronal cells treated with Aβ 25 - 35 exhibited mitochondrial calcium overload, which was associated with ATP depletion, ROS generation, permeability transition pore opening, caspase-9 activation, and cell death. Sildenafil prevented these effects by promoting the opening of ATP-sensitive K + channels [140]. In cultured HT-22 hippocampal neuronal cells, exposure to advanced glycation end products [141] (a risk factor for AD [142]) induced mitochondrial ROS generation, depleted intracellular ATP, opened the mitochondrial permeability transition pore, released cytochrome C, activated caspase-3, and initiated apoptosis. Treatment with sildenafil upregulated heme oxygenase 1 (HO1), protected mitochondria from permeability transition pore opening and cytochrome C release, and decreased caspase-3 activation and apoptosis [141]. HO1 expression was required for sildenafil-induced protection of mitochondrial reductive capacity and permeability transition pore opening [141], suggesting that sildenafil protected mitochondria via HO1 upregulation.
In mice with cholinergic dysfunction mimicking AD due to scopolamine injection, sildenafil rescued maze performance, with 3 mg/kg appearing to be more efficacious than the 1.5 or 4.5 mg/kg dosages [143].
In hippocampal slices from transgenic amyloid precursor protein (APP)/presenilin 1 (PS1) AD mice, 50 nM sildenafil rescued the deficits in tetanus-induced long-term potentiation in the Schaffer collateral pathway caused by the APP/PS1 genotype [144]. In vivo sildenafil treatment produced similar results. In APP/PS1 mice, a one-time dosage of 3 mg/kg sildenafil rescued contextual fear memory. Daily intraperitoneal dosages for 2-3 weeks of 3 mg/kg sildenafil partially rescued spatial working memory deficits on the radial arm water maze test. Similar benefits were found for daily dosages of 3 mg/kg sildenafil 9–12 weeks after treatment cessation, indicating a long-term benefit that persists even after treatment cessation. Sildenafil also rescued long-term spatial reference memory on the Morris water maze and the probe trial. Sildenafil treatment rescued tetanus-induced CREB phosphorylation in the CA1 hippocampus to normal levels. 3 weeks of daily 3 mg/kg sildenafil was sufficient to reduce Aβ 40 and Aβ 42 levels in cerebral cortex samples [144].
In APP/PS1 mice administered sildenafil 6 mg/kg intraperitoneally daily for 3 months, significant improvements were documented in behavioral tests (nesting behavior, arm entries in the Y maze, Morris water maze escape latency and path length), as well as immunoreactivity of inflammatory microglial and astrocytic markers ionized calcium binding adaptor molecule 1 (Iba1) and glial fibrillary acidic protein (GFAP), neurogenesis as shown by NeuN-positive neurons and doublecortin (DCX)-positive cells in dentate gyrus, and amyloid plaque burden [145].
In APP/PS1 AD mice, 2 mg/kg sildenafil twice daily for 4 months rescued cognition as shown by spontaneous alternation and escape from electrical stimulation in the Y-maze test, decreased amyloid pathology as shown by decreased cortical and hippocampal AβPP, Aβ 40, and Aβ 42 levels, decreased PDE5 expression, and increased nNOS, eNOS, iNOS, NO, and cGMP levels [146].
Sub-chronic intraperitoneal sildenafil treatment in APP/PS1 mice was found to improve memory as shown by novel object recognition preference, downregulated proinflammatory cytokines interleukin-1β (IL-1β), intereukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) in the hippocampus, decreased hippocampal soluble Aβ 40 and Aβ 42 expression, and increased CREB phosphorylation [147].
A recent systematic review found that, on the Morris water maze and the T-maze, sildenafil improved spatial memory retention but not acquisition in aged mice [64]. Interestingly, this review reported that PDE5 is not located in brain structures critical for AD based on the lack of specific hybridization of a PDE5 mRNA probe [62–64], but as noted, other groups have found increased PDE5 mRNA or protein expression in AD patients’ entorhinal [58] or temporal cortices [55], respectively, and PDE5 mRNA expression has been found by multiple groups in the normal human brain, albeit at lower levels than in peripheral tissues [58–61].
In J20 AD mice, 15 mg/kg sildenafil daily for 10 weeks in drinking water resulted in improved performance on the Morris water maze, decreased tau hyperphosphorylation, and increased Akt and GSK3β phosphorylation, but it did not alter prefrontal cortex Aβ 42 levels [61].
In Tg2576 AD mice, 15 mg/kg/day intraperitoneal sildenafil significantly rescued learning and memory deficits as shown by the Morris water maze and fear conditioning tasks, reduced hippocampal tau hyperphosphorylation, GSK3β activity, and the CDK5 p25/p35 ratio, upregulated hippocampal p-CREB and c-Fos following fear conditioning training, and increased hippocampal expression of brain-derived neurotrophic factor (BDNF) and activity-regulated cytoskeletal-associated protein (Arc) (an immediate early response gene involved in memory encoding) [148]. However, sildenafil did not affect total Aβ 42 levels in the frontal cortex [148].
In aged mice, 3 mg/kg intraperitoneal sildenafil daily for 3 weeks decreased double-stranded DNA breaks and apoptotic cells as visualized by the terminal deoxyuridine triphosphate nick end labeling (TUNEL) assay in the CA1 hippocampus, downregulated proapoptotic proteins caspase-3 and B-cell lymphoma 2-associated X (Bax), upregulated antiapoptotic B-cell lymphoma protein-2 (Bcl2) and BDNF, downregulated AβPP expression, and suppressed the age-associated shift in the Aβ 42/Aβ 40 ratio [149].
In the senescence accelerated mouse-prone 8 (SAMP8) mouse model of accelerated aging and sporadic AD, 7.5 mg/kg sildenafil for 4 weeks improved cognitive performance as shown by the Morris water maze and the passive avoidance test [145, 151], tau hyperphosphorylation [145, 151], inflammation as shown by GFAP downregulation, and amyloid pathology as shown by downregulation of BACE1, cathepsin B, and hippocampal Aβ 42 [150]. Sildenafil also activated Akt and inhibited GSK3β, calpain, cyclin-dependent kinase 5 (CDK5) [145, 150], and c-Jun N-terminal kinase (JNK) [151].
Sildenafil in AD patients
In 10 AD patients, a single 50 mg dose of sildenafil significantly decreased spontaneous neural activity in the right hippocampus as shown by the fractional amplitude of low-frequency fluctuations recorded on functional magnetic resonance imaging of the blood oxygen level-dependent signal, a parameter that had been shown to be aberrantly increased in AD patients’ hippocampi and parahippocampal gyri [152]. In 12 elderly patients with AD, a single dosage of 50 mg of sildenafil significantly increased the cerebral metabolic rate of oxygen and cerebral blood flow [127]. In 8 AD patients, it decreased cerebrovascular reactivity [127].
DISCUSSION
As predicted in the introduction, preclinical studies that tested these parameters have found that sildenafil rescued CREB phosphorylation, long-term potentiation, and learning and memory [61, 151], increased neurogenesis [145], and decreased neuroinflammation [145, 150]. In addition, these studies consistently found that sildenafil decreased tau hyperphosphorylation and related parameters [61, 151]. This might be in part because the MnSOD downregulation in AD hippocampal neurons contributes to tau hyperphosphorylation [45,153, 45,153], and low-dose sildenafil appears to upregulate MnSOD via PGC1α activation [69–72]. The relatively high 15 mg/kg dosages appear to have resulted in decreased tau hyperphosphorylation predominantly because high-dose sildenafil activated the cGMP/PKG/PI3K/Akt pathway, leading to increased inhibitory Ser9 phosphorylation of tau kinase GSK3β [61, 148].
Discrepancies have been reported, however, regarding the effect of sildenafil on Aβ levels: most of the studies showed that sildenafil decreases Aβ levels [144–147, 150], but the two studies using the highest dosages (15 mg/kg) reported no effect on frontal cortex Aβ levels [61, 148]. This can be understood through the lens of the U-shaped dose-response curve of sildenafil and cGMP on PGC1α signaling documented in the introduction: low-dose sildenafil and cGMP appear to activate PGC1α and suppress BACE1 expression [65, 76], whereas high-dose sildenafil and 100μM cGMP appear to inhibit PGC1α due to crosstalk with cAMP and PDE2 signaling, failing to suppress BACE1 expression. In other words, it is possible that the 15 mg/kg sildenafil studies may not have decreased Aβ levels significantly [61, 148] because that dosage activates PDE2, depletes cAMP [76], and inhibits cAMP-signaled PGC1α activation and BACE1 repression [65, 67]. Another intriguing observation is that only the 15 mg/kg studies reported Akt activation and/or GSK3β inhibition [61, 148], suggesting the possibility that the 15 mg/kg dosages activated Akt [61, 148] and may have consequently repressed PGC1α expression and its anti-amyloidogenic properties (Fig. 4) [80–82].
Interestingly, in vitro studies found that sildenafil protected mitochondria from Aβ or AGEs via ATP-sensitive K + channels or HO1 upregulation, respectively [140, 141], suggesting that sildenafil may promote mitochondrial function via multiple mechanisms, some of which may be independent of PGC1α.
In patients, an especially promising finding is that 50 mg sildenafil increased the cerebral metabolic rate of oxygen [127]. This effect might be accounted for totally by the increases in the cerebral blood flow [127], or it may have been partially mediated as well by PGC1α-regulated mitochondrial biogenesis. However, none of the studies reviewed measured PGC1α mRNA, protein, or acetylation levels, nor other markers of mitochondrial biogenesis, making it impossible to evaluate this possibility. Nor was the effect of sildenafil on SIRT1 activity or the SIRT1/PGC1α pathway explored in any of these studies. Nor did these studies examine the effect of sildenafil on markers of insulin resistance or antioxidant enzyme expression. Future preclinical studies in transgenic AD mice should address these points directly to assess the possible role of SIRT1 and PGC1α activity in sildenafil-induced Aβ suppression, mitochondrial biogenesis, and antioxidant enzyme expression in AD, as well as the putative effect of sildenafil on insulin resistance. Future studies should also assess the potential of combining sildenafil with a PDE2 inhibitor to increase sildenafil’s maximal effective dosage continuously throughout the treatment duration. This would bypass the dose-limiting effect of sildenafil on PDE2 activation and cAMP depletion [76], allowing for robust simultaneous cAMP and cGMP signaling and therefore maximal PGC1α activity, mitochondrial biogenesis, antioxidant enzyme expression, and BACE1 repression [48, 154–156]. The best candidate for this role would be propentofylline, a potent and broad-spectrum PDE inhibitor and methyl xanthine derivate like caffeine that is particularly effective at inhibiting cGMP-stimulated PDE2 activity and PDE4 [157]. Propentofylline would be superior to other PDE2 inhibitors primarily because 300 mg of it taken thrice daily one hour before meals has been tested and found to be safe and effective in mild to moderate AD patients in 5 phase III clinical trials [158–164]. Intriguingly, a recent review by Heckman and colleagues opined that, based on the preclinical evidence, inhibition of PDE2, PDE4, and PDE5 seemed to hold the most promise for the treatment of AD [165], and sildenafil and propentofylline administered together would potently inhibit these three therapeutic targets simultaneously [157].
Ultimately, a randomized control trial of sildenafil should be undertaken in AD patients to assess the clinical benefits of long-term sildenafil administration in this population compared to elderly controls. This RCT should use the 50 mg/day dosage [127]. As outcome measures, the RCT should test cognition on the MMSE and the Alzheimer’s Disease Assessment Scale-Cognitive Subscale, comorbid depression on the Geriatric Depression Scale [56], amyloid and tau pathology binding with 2-(1-6-[(2-[F- 18]fluoroethyl)(methyl)amino]-2-naphthylethylidene)malononitrile positron emission tomography (FDDNP-PET) [166, 167], cerebral blood flow with MRI [127], the cerebral metabolic rate of oxygen with MRI [127, 169], the cerebral metabolic rate of glucose with 18F-fluoro-deoxyglucose positron emission tomography (FDG-PET) [34, 170–174], inflammation with CSF IL-1β, IL-6, and TNF-α [124–126, 175–182], NOS/NO/sGC/cGMP/PGC1α pathway dysfunction with CSF cGMP [55, 56], and antioxidant enzyme activity with urine 8-oxo-2’-deoxyguanosine as an indirect biomarker [45, 183].
CONFLICT OF INTEREST
The authors have no conflict of interest to report.