
Abstract
Background:
Preclinical studies, clinical trials, and reviews suggest increasing 3’,5’-cyclic adenosine monophosphate (cAMP) and 3’,5’-cyclic guanosine monophosphate (cGMP) with phosphodiesterase inhibitors is disease-modifying in Alzheimer’s disease (AD). cAMP/protein kinase A (PKA) and cGMP/protein kinase G (PKG) signaling are disrupted in AD. cAMP/PKA and cGMP/PKG activate cAMP response element binding protein (CREB). CREB binds mitochondrial and nuclear DNA, inducing synaptogenesis, memory, and neuronal survival gene (e.g., brain-derived neurotrophic factor) and peroxisome proliferator-activated receptor-γ coactivator-1α (PGC1α). cAMP/PKA and cGMP/PKG activate Sirtuin-1, which activates PGC1α. PGC1α induces mitochondrial biogenesis and antioxidant genes (e.g.,Nrf2) and represses BACE1. cAMP and cGMP inhibit BACE1-inducing NFκB and tau-phosphorylating GSK3β.
Objective and Methods:
We review efficacy-testing clinical trials, epidemiology, and meta-analyses to critically investigate whether phosphodiesteraseinhibitors prevent or treat AD.
Results:
Caffeine and cilostazol may lower AD risk. Denbufylline and sildenafil clinical trials are promising but preliminary and inconclusive. PF-04447943 and BI 409,306 are ineffective. Vinpocetine, cilostazol, and nicergoline trials are mixed. Deprenyl/selegiline trials show only short-term benefits. Broad-spectrum phosphodiesterase inhibitor propentofylline has been shown in five phase III trials to improve cognition, dementia severity, activities of daily living, and global assessment in mild-to-moderate AD patients on multiple scales, including the ADAS-Cogand the CIBIC-Plus in an 18-month phase III clinical trial. However, two books claimed based on a MedScape article an 18-month phase III trial failed, so propentofylline was discontinued. Now, propentofylline is used to treat canine cognitive dysfunction, which, like AD, involves age-associated wild-type Aβ deposition.
Conclusion:
Phosphodiesterase inhibitors may prevent and treat AD.
Keywords
BACKGROUND
Sporadic Alzheimer’s disease (AD) is the most common cause of dementia in the elderly. It involves brain amyloid-β (Aβ) generation, aggregation into oligomers, and deposition into neuritic plaques, tau hyperphosphorylation into neurofibrillary tangles [1, 2], iron and aluminum accumulation [3, 4], infections [5–11], oxidative stress and damage to lipids, RNA, DNA, and proteins [12–16], aberrant calcium and zinc signaling [17–21], mitochondrial dysfunction [22–24],endoplasmic reticulum stress [25–27], lysosomal dysfunction [17, 28–30], defective autophagy [31], neuroinflammation [32–38], neuronal abortive cellcycle re-entry [39–48], insulin and insulin-like growth factor 1 resistance [49–51], synaptic dysfunction [52], and neuron death.
Cyclic nucleotides are endogenous cell signaling molecules that act as second messengers to regulate various physiological processes in the central nervous system. There are multiple cyclic nucleotides that may be involved in the pathogenesis of AD, including Ca2+-mobilizing and transient receptor potential melastatin 2 (TRPM2) channel-openingcyclic adenine diphsophate ribose (cADPR) [53–57] and cytoplasmic DNA-induced cyclic guanosine monophosphate adenosine monophosphate (cyclic GMP-AMP, or cGAMP)[14–16, 58–62]. Of these though, there is the most evidence about the cyclic purine nucleotides 3’,5’-cyclic adenosine monophosphate (cAMP) and 3’,5’-cyclic guanosine monophosphate (cGMP).
Physiologic cAMP and cGMP signaling
Physiologically, cAMP is produced whenGαs-containing complexes activate adenylate cyclases (AC). Adenylate cyclase generates cAMP from adenosine triphosphate (ATP). cAMP activates exchange protein directly activated by cAMP (EPAC) and protein kinase A (PKA). PKA holoenzymes consist of two catalytic and two regulatory subunits. When bound to cAMP, the regulatory subunits release the catalytic PKA subunits to phosphorylate many targets [63].
Regarding cGMP signaling, nitric oxide synthases (NOS) produce nitric oxide (NO).NO activates soluble guanylyl cyclase (sGC) in the cytoplasm. sGC produces cytoplasmic cGMP [64, 65]. In addition, Atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP) activate particulate, plasma-membrane localized, receptor guanylyl cyclase-A (GC-A) [66], and C-type natriuretic peptide (CNP) activates the particulate guanylyl cyclase B (GC-B) [67]. Particulate GC-A and GC-B produce cGMP proximal to the plasma membrane [67, 68]. cGMP activates protein kinase G (PKG) [64, 65].
Phosphodiesterases
Phosphodiesterase (PDE) enzymes degrade cyclic purine nucleotides. PDE1, 2, 3, 10, and 11 degrade both cAMP and cGMP [69, 70]. PDE4, 7, and 8 degrade cAMP specifically. PDE5, 6, and 9 degrade cGMP specifically [69, 70]. PDE5 degrades cytoplasmic cGMP produced by NOS/NO/sGC signaling, whereas PDE9 degrades peri-plasma membrane cGMP produced by natriuretic peptide/pGC signaling [71]. Complicating the interpretation of studies about the effects of cGMP, low concentrations of cGMP inhibit PDE3, increasing cAMP levels, whereas moderately high doses of cGMP activate PDE2, decreasing cAMP levels [72].
Various drugs and naturally-occurring compounds inhibit one or more PDE enzyme. Based on the various preclinical studies about PDE inhibition for AD, a review by Heckman et al. concluded that PDE2, PDE4, and PDE5 inhibitors appear to hold the most promise for treating AD [73]. Multiple benefits of increasing cAMP and/or cGMP signaling with various PDE inhibitors can be ascribed to the effect of these pathways on cAMP response element binding protein (CREB), Sirtuin 1 (SIRT1), peroxisome proliferator-activated receptor-γ coactivator-1α (PGC1α), nuclear factor erythroid 2-related factor 2 (Nrf2), nuclear factor κ-light-chain-enhancer of activated B cells (NFκB), and glycogen synthase kinase 3β (GSK3β) signaling (Fig. 1). The following section is a discussion of the effects of cAMP and cGMP levels on these molecular pathways.
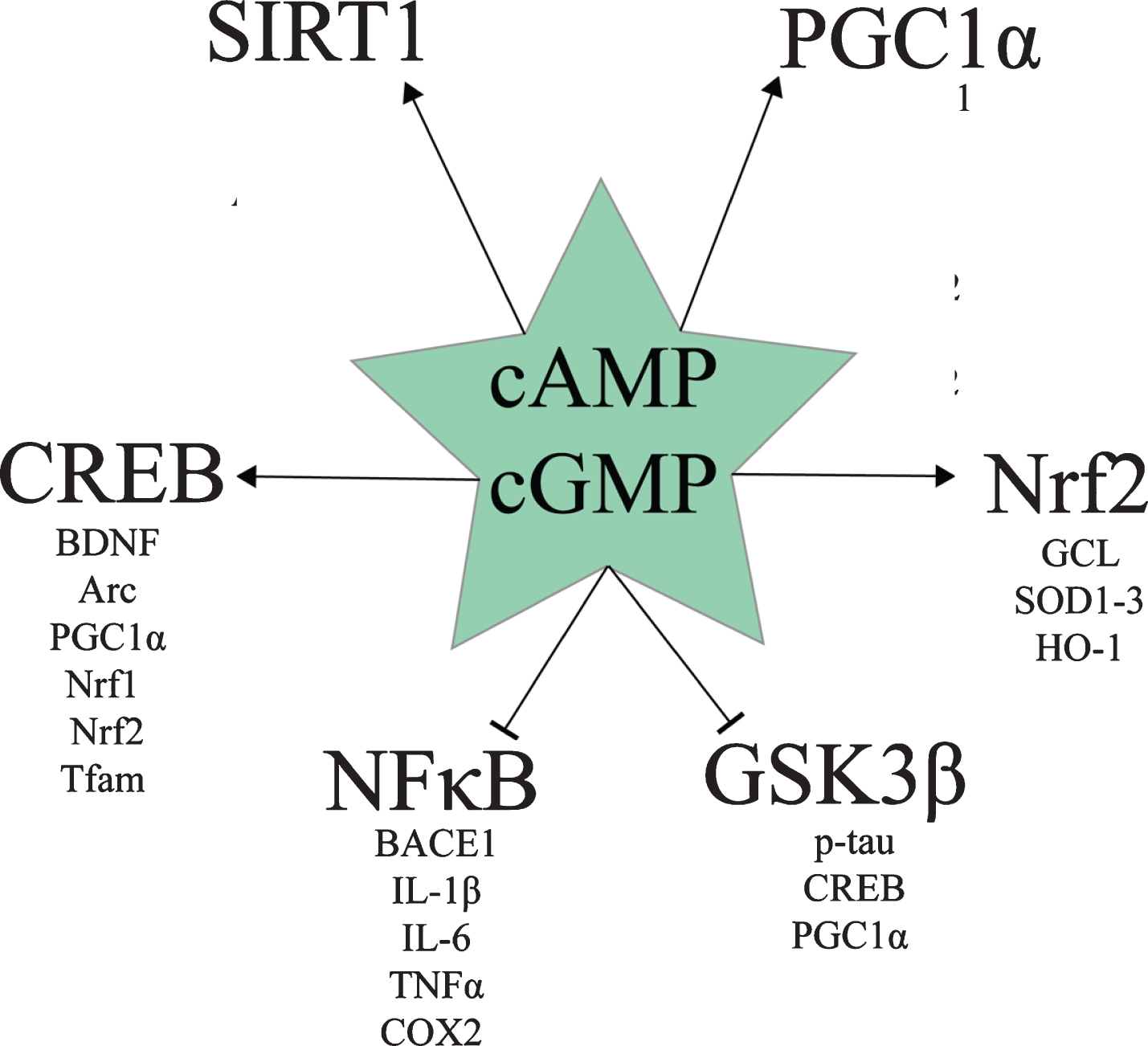
Downstream targets of cAMP and cGMP signaling relevant to AD.
cAMP, cGMP, and CREB
Both cAMP/PKA and cGMP/PKG signaling activate CREB by phosphorylating it at Ser133 [74–79]. pSer133-CREB protein levels are decreased in the AD hippocampus [80], the AD prefrontal cortex, and AD peripheral blood mononuclear cells [81]. pSr133-CREB activates the transcription of mitochondrial D-loop DNA and nuclear DNA [82, 83]. Activated CREB induces genes involved in synaptogenesis and memory formation [84], such as brain derived neurotrophic factor (BDNF) and Activity-regulated cytoskeleton-associated protein (Arc), and neuronal survival, such as BDNF and Bcl2 [84–90], as well as genes involved in mitochondrial biogenesis and antioxidant defense, such as PGC1α, Nuclear respiratory factor 1 (Nrf1), Nrf2, and mitochondrial transcription factor A (Tfam) [83, 91].
cAMP, cGMP, SIRT1, and PGC1α
Both cAMP/PKA and cGMP/PKG signaling promote mitochondrial biogenesis and antioxidant gene expression via CREB- and SIRT1-mediated PGC1α activation [92–105]. In addition to activating CREB [74–79], which induces PGC1α transcription [83, 91], both cAMP/PKA and cGMP/PKG signaling activate SIRT1 [94–96, 104–110]. SIRT1 exerts multiple beneficial effects in AD, including downregulating pro-amyloidogenic β-secretase (BACE1) and upregulating anti-amyloidogenic A Disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) [106], as well as deacetylating and thereby post-translationally activating PGC1α [97, 111–118].
PGC1α induces the core regulators of mitochondrial biogenesis, including Nrf1, Nrf2, and Tfam [93]. PGC1α induces antioxidant genes when bound to Forkhead box O3 (Foxo3a) and deacetylated by SIRT1, namely manganese superoxide dismutase (MnSOD), catalase, peroxiredoxins 3 and 5, thioredoxin 2, thioredoxin reductase 2, and uncoupling protein 2 (UCP2)[112].Brain PGC1α overexpression has also been shown to upregulate glutathione peroxidase 1 (GPx1), UCP4, and UCP5 [119]. PGC1α also induces genes involved in oxidative respiration [92, 103] and fatty acid β-oxidation [120]. Conversely, in a peroxisome proliferator-activated receptor γ (PPARγ)-dependent mechanism, PGC1α represses the expression of BACE1 [121, 122].
These downstream effects of PGC1α signaling are important for AD treatment because BACE1 is the rate-limiting enzyme in Aβ generation [123], and neural mitochondrial biogenesis and antioxidant enzyme expression are severely disrupted in AD. In the AD patient hippocampus, mitochondrial biogenesis is impaired as shown by decreased number of mitochondria in neurons and lower expression of PGC1α, as well as of downstream Nrf1, Nrf2, Tfam [93], and MnSOD [23, 112]. This deficit in PGC1α signaling in AD appears to begin very early. PGC1α, Nrf1, Nrf2, and Tfam were downregulated at the mRNA level as early as 1 month of age in a transgenic AD mouse model, even before a significant concentration of Aβ oligomers had formed [91]. In rodent brain hypo-perfused like AD and vascular dementia brains are [119, 124–127], PGC1α neural overexpression improved cognitive deficits and the metabolic activity of hypoxic neurons [119], indicating the therapeutic potential of modulating PGC1α signaling pharmacologically.
Therapeutically, PDE inhibitorsprovide a way to modulate this AD deficit in pSer133-CREB and PGC1α signaling. In APPswe M17 cells, cAMP rescued CREB phosphorylation and PGC1α expression in a PKA-dependent manner [93]. Furthermore, cAMP signaling may promote not only synaptogenesis, memory formation, neuron survival, and mitochondrial biogenesis, but also restoration of the physiologic balance between mitochondrial biogenesis and mitophagy. For example, 15 mg/kg/day of the cAMP-elevating PDE7 inhibitor S14 given orally to 6-month-old APP/PS1 mice rescued memory deficits, increased neurogenesis, and rescued mitochondrial mass by increasing mitochondrial biogenesis and decreasing Aβ-induced excessive mitophagy [128]. This balancebetween mitochondrial biogenesis and mitophagy appears to be disrupted in AD patients’ hippocampal pyramidal neurons as shown by the PGC1αdownregulation [93, 129], as well as the finding that many of the few surviving mitochondria accumulate in autophagosomes [22, 130]. Ultimately, in hypoperfused rodent brain, the nonspecific PDE inhibitor propentofylline restores mitochondrial ATP production [131, 132], suggesting that one outcome of modulating CREB and PGC1α signaling with PDE inhibitors is enhanced mitochondrial ATP production.
cAMP, cGMP, and Nrf2
Downstream of CREB and PGC1α, Nrf2 is a transcription factor that induces various genes involved in antioxidant defense, detoxification, autophagy, and cytoprotection, such as glutamate–cysteine ligase (the rate-limiting enzyme in glutathione synthesis), SOD1-3, and heme oxygenase 1 (HO-1) [133–136]. Nrf2 is active when it is localized to the nucleus; however, nuclear Nrf2 levels are decreased in AD patients’ hippocampal cells, indicating decreased Nrf2 activity in AD [137]. Both cAMP and cGMP appear to increase Nrf2 signaling.
cAMP appears to increase Nrf2 signaling in most cell types [138–140]. For example, in mouse and human hepatocytes, cAMP analogue 8-Bromoadenosine-cAMP upregulated Nrf2 target gene expression and antioxidant response element (ARE) transcriptional activity in a PKA-dependent manner [138]. Treatment of keratinocytes and melanocytes with either α-melanocyte stimulating hormone or forskolin (both of which increase cAMP) increased Nrf2 and Nrf2 target gene expression [139]. The PDE4 inhibitor roflumilast has been shown to replenish Nrf2 levels in bronchial epithelial cells infected with respiratory syncytial virus [141, 142]. Therefore, cAMP may increase Nrf2 signaling in neurons and other brain cells [138–140].
cGMP may also activate Nrf2 signaling, as in human bronchial epithelial cells, arsenite activated Nrf2 in a cGMP/PKG-dependent manner [143]. cGMP/PKG signaling has been postulated to be involved in Nrf2 activation in the heart as well [144]. In C6 glioma cells, nitrated cGMP activated Nrf2 [145]. In colon carcinoma cells, nitric oxide activated Nrf2 [146]. This points to context-dependent activation of Nrf2 by cGMP, nitrated cGMP, and NO in diverse cell types. Therefore, both cAMP and cGMP (and thus PDE inhibitors) may promote Nrf2 activity in brain cells.
cAMP, cGMP, and NFκB
Another important therapeutic target in AD is NFκB. NFκB is a family of transcription factor dimers consisting of two of five possible protein subunits: c-Rel, RelA/p65, RelB, p105/NFKB1, and p100/NFKB2. NFκB dimers containing the RelA/p65 subunit induce inflammatory gene expression (e.g., pro-inflammatory cytokines IL-1β, IL-6, and TNFα, chemokines IL-8, RANTES, MIP1, enzymes COX2 and iNOS, adhesion molecules VCAM1 and ICAM1 [147]), as well as the expression of BACE1 [34, 35]. Inflammatory NFκB is activated by oxidative stress [148], diverse pathogen- and damage-associated molecular patterns via toll-like receptors (TLR) [149, 150], fibrillar Aβ via TLR2 [32], pro-inflammatory cytokines (e.g., TNFα via the TNF receptor and IL-1β via the IL-1 receptor) [147], and other immunoreceptors [37, 148], such as the B-cell antigen receptor (BCR) in conjunction with protein tyrosine kinases [151]. Through signaling mediators such as MYD88 [32], TLR, for example, activate inhibitor of NFκB kinase (IKK), which phosphorylates inhibitor of NFκB protein α (IκBα), causing its ubiquitination and degradation, releasing c-Rel, RelA/p65, and RelB containing dimers to translocate to the nucleus and induce gene transcription [151].
cAMP/PKA signaling inhibits NFκB in most but not all cell types and contexts [147, 152]. With relevance to AD, raising cAMP has been shown to inhibit NFκB in microglia stimulated with either TNFα or lipopolysaccharide (LPS) [147, 154]. Disruption of inflammatory NFκB signaling by cAMP has been shown to occur via multiple mechanisms [147, 155]. Specifically, cAMP/PKA-phosphorylated CREB upregulates the IκBα gene [147, 157]. cAMP inhibits IKKβ, decreasing IκB phosphorylation, ubiquitination, and degradation [147, 157]. cAMP inhibits the ubiquitination and degradation of IκBα [147, 151]. Phosphorylated CREB outcompetes NFκB for their common requisite binding partner CREB binding protein (CBP), shifting gene transcription towards CREB and away from NFκB-mediated expression [147]. cAMP induces the exchange of activating p65-p50 NFκB dimers for repressive p50-p50 dimers [147]. cAMP induces the exchange of activating CREB-c-Jun dimers for repressive CREB-ICER dimers in the promoters of certain genes, such as TNFα [147]. cAMP activates the expression of c-Fos, which prevents p65 homodimers from binding to promoters [147].
cGMP inhibits NFκB indirectly via cAMP [155, 158]. cGMP inhibits PDE3, raising cAMP levels. In vascular smooth muscle cells, raising effective cGMP levels with a NO donor or C natriuretic peptide resulted in cGMP-induced PDE3 inhibition, cAMP accumulation, and cAMP/PKA-dependent NFκB inhibition [158].
Therefore, raising cAMP and cGMP levels with PDE inhibitors would likely inhibit NFκB in brain microglia, decreasing neuroinflammation and β-amyloidosis in AD [34, 156–158].
cAMP, cGMP, and GSK3β
GSK3β is the primary kinase of 45 of the 85 phosphorylable Serine and Threonine residues on tau [159–162]. If PKA phosphorylates tau before GSK3β does, then paired helical filament formation is suppressed, whereas if GSK3β phosphorylates tau before PKA does, then paired helical formation is promoted [163], suggesting that high PKA activity and low GSK3β activity may suppress neurofibrillary tangle formation. Furthermore, GSK3β phosphorylates and thereby inhibits CREB and PGC1α [101, 164–169].
Multiple lines of evidence suggest that both cAMP and cGMP inhibit GSK3β. cAMP/PKA suppresses GSK3β activity in most cell types by phosphorylating it at Ser9, including in cortical neurons [170–172], platelets [173], mouse spermatozoa [174], Rat1, NIH 3T3, and HEK293 cells [175], but not in murine melanoma cells [176]. Multiple AD rodent model studies have shown that the cGMP-elevating PDE5 inhibitor sildenafil inhibits GSK3β, suggesting that cGMP typically inhibits GSK3β in the brain [177–181].
Gαs/AC/cAMP/PKA signaling alterations in AD
The beneficial effects of cAMP and cGMP on CREB, PGC1α, Nrf2, NFκB, and GSK3β signaling, however, appear to be lost during the pathogenesis of AD, as preclinical studies and neuropathology evidence suggestthat the AC/cAMP/PKA and NOS/NO/sGC/cGMP pathways may be pathologically disrupted in AD patients’ brains [79, 182–205]. In preclinical models, both BACE1/Aβ and tau pathology disrupt cAMP signaling. Aβ25–35 exposure transiently increases cAMP levels, suppressing neuronal glucose uptake in a cAMP/PKA-dependent manner [206], but long-term Aβ42 exposure decreases cAMP levels [182]. Sublethal doses of Aβ42 (but not Aβ25–35) impair KCl- and N-methyl-D-aspartate-induced CREB phosphorylation [193]. BACE1 overexpression decreases cAMP levels, PKA activity, and CREB phosphorylation independently of Aβ [199]. Human tau overexpression inhibits PKA, decreases phosphorylation of CREB, decreases phosphorylation of glutamate receptor 1 [200] (GluA1, phosphorylation of which increases its membrane localization, facilitating conductivity, long-term potentiation, and memory formation [207]), decreases phosphorylation of tropomyosin receptor kinase B [200] (TrkB, a receptor for BDNF [208]), and downregulates BDNF mRNA and protein levels [200]. Therefore, both Aβ and tau pathology appear to disrupt cAMP/PKA signaling.
In neuropathology studies, it appears that AC/cAMP/PKA signaling may be disrupted in AD patients’ brains. To begin with, AC protein and activity levels (especially Gαs-stimulated) have generally been found to be decreased in AD patients’ brains [183–187, 201–205]. Basal, Gαs-stimulated, and forskolin-stimulated AC activity has been found to be decreased in the AD hippocampus [201–203]. However, Gαs-stimulated but not basal or forskolin-stimulated AC activity was found to be decreased in the AD hippocampus, temporal cortex, and angular gyrus in one study [204]. AC-I protein levels were significantly decreased in the AD hippocampus [205]. Parietal cortex membrane AC-I and AC-II protein levels were significantly decreased in AD patients, as was Ca2 + /CaM-stimulated AC activity [183]. Basal AC activity was found to be decreased in the AD frontal cortex, temporal cortex, and angular gyrus [184]. Gαs-stimulated but not basal or forskolin-stimulated AC activity was decreased in the AD superior temporal cortex [185]. Gαs-stimulated but not basal or forskolin-stimulated AC activity was decreased in the AD neocortex [186]. [3H]forskolin binding was decreased in the AD frontal cortex, suggestive of fewer AC catalytic subunits to bind to [187]. These findings overall suggest that a decrease in AC activity may occur in AD brains.
cAMP levels were not altered in the AD patient cerebrospinal fluid (CSF)in two studies [197, 198], but they were elevated in one study in a manner that correlated with tau pathology [209]. Increased cAMP levels were found in AD microvessels [189], and in the cerebral cortical and meningeal vessels in the frontal and temporal cortex and hippocampus in association with vascular Aβ [188]. This suggests that cAMP levels and localization may be subtly dysregulated in AD brains.
Downstream of cAMP, PKA activity appears to be suppressed in AD in most studies [185, 211]. [3H]cAMP binding to cytosolic but not particulate PKA was decreased in the AD patient entorhinal cortex and subiculum [190]. Neither soluble nor particulate PKA activity were altered in the AD patient superior temporal cortex in one study [185], nor was PKA activity altered in AD patient cerebral microvessels [210]. In another, however, decreased PKA activity was found in the AD patient temporal cortex [192]. Decreased PKA activity was also observed in the AD patient medial temporal cortex [191]. Regulatory 51 kDa PKA-RIIα and PKA-RIIβ and catalytic PKA-Cβ protein levels were decreased in the AD patient medial temporal cortex [191]. Catalytic PKA-Cα protein levels were decreased in the AD patient frontal cortex [194]. However, a calpain-cleaved 47 kDa fragment of PKA-RIIα was increased in the AD patient medial temporal cortex [191]. In cultured hippocampal neurons, Aβ exposure resulted in PKA inhibition, higher PKA-RIIα protein levels, and less glutamate-induced CREB phosphorylation (the latter of which was reversed by cAMP-elevating PDE4 inhibitor rolipram) [211]. Overexpression of human tau was found to increase nuclear PKA-RIIα proteins [200], suggesting that the concerted action of Ca2+-activated calpain proteases, Aβ, and tau may promote expression of the 47 kDa PKA-RIIα fragment in the nucleus. Silencing PKA-RIIα expression improved tau-induced deficits in PKA activity and CREB phosphorylation [200], suggesting that this nuclear 47 kDa PKA-RIIα upregulation in AD may suppress nuclear PKA activity and CREB phosphorylation. Therefore, overall PKA activity and nuclear PKA activity appear to be suppressed in AD [185, 211].
GC/cGMP/PKG signaling alterations in AD
Preclinical and neuropathology data suggest that NOS/NO/sGC/cGMP signaling may be disrupted in AD as well. Aβ inhibits the NOS/NO/sGC/cGMP/CREB pathway required for long-term potentiation in hippocampal slices [79]. NOS activity has been found to be decreased in the AD superior frontal gyrus and hippocampus [196]. The activity of NO-activated sGC—but not basal sGC nor particulate GC (pGC)—was found to be decreased in the AD superior temporal cortex [195].
Upstream of the maintained pGC signaling [195], elevated BNP levels have been associated with cognitive disorders in elderly patients 75–78 years old but not older [212, 213], and multiple studies have found associations between increased BNP levels in blood or plasma andmild cognitive impairment (MCI) occurrence, severity, or risk, conversion of MCI to AD, or dementia [213–223]. For example, one study found plasma BNP levels were associated with diagnosis of MCI or AD as well as CSF Aβ42 levels; furthermore, plasma BNP levels were found to be increased in MCI and AD patients [213, 214]. This suggests that BNP/pGC/cGMP signaling may be either maintained or increased in MCI and AD [195, 212–214].
Overall, cGMP levels were found to be decreased in AD patient CSF [197, 198], which correlated with CSF levels of Aβ42 [197], depression symptoms [198], and cognitive dysfunction as measured by The Mini-Mental Status Exam (MMSE) [197, 198]. This suggests that AD is characterized by a profound and disease severity-correlating decrement in NOS/NO/sGC/cGMP signaling.
Increasing cAMP and cGMP signaling to treat AD
Thus, PDE inhibitors would be predicted to restore disrupted cAMP/PKA, cGMP/PKG, CREB, PGC1α, and Nrf2 signaling, activate SIRT1, repress NFκB-induced BACE1 expression, Aβ generation, and inflammation,inhibit GSK3β-mediated tau phosphorylation,and promote neuron survival, synaptogenesis, mitochondrial biogenesis and survival of mitophagy, ATP generation, antioxidant and detoxification enzyme expression, and memory formation—all of which would be crucial therapeutic benefits in the fight to prevent, halt, and reverse AD etiopathogenesis. In this narrative review, we overview efficacy-testing clinical trials, epidemiological studies, and meta-analyses to investigate whether PDE inhibitors can prevent or treat AD, MCI, or dementia in a disease-modifying fashion.
CLINICAL TRIALS AND EPIDEMIOLOGY OF PDE INHIBITORS
Vinpocetine
Vinpocetine inhibits PDE1, raising cAMP and cGMP [224]. Discovered in the 1970s by C. Szántay and C. Lörincz et al. independently, vinpocetine has been approved in European countries for the treatment of dementia and stroke for over 30 years, and it is available in the United States as a dietary supplement [225]. Preclinical data demonstrate that vinpocetine can rescue cognitive deficits in a rodent AD model [226], increase CREB phosphorylation [227] and BDNF expression [228], downregulate BACE1 [226], upregulate Nrf2 mRNA expression [229], decrease oxidative stress [226, 228–239], mitochondrial dysfunction [240, 241], and apoptosis [230, 239], inhibit GSK3β [226, 228], NFκB [229, 242–250], and the NLPR3 inflammasome [243], decrease levels of pro-inflammatory cytokines IL-1β [226, 243], IL-6 [236, 251], and TNFα [226, 246], and arrest the cell cycle in the G1 phase by downregulating cyclin D1 and upregulating p27(Kip1) [252].
Despite these promising preclinical findings, there is very little evidence about vinpocetine for the treatment of AD specifically, and what little there is is disappointing. In a 1-year-long 1989 open-label pilot trial, 15 patients with AD showed no improvement or delay in decline on the CGI at doses of 30, 45, or 60 mg vinpocetine once daily [253]. In a multicenter, double-blind, placebo-controlled trial, subgroup analysis showed that no significant treatment effect was observed on the CGI, the Short Cognitive Performance Test/Syndrom-Kurztest (SKT), the Brief Cognitive Rating Scale, or the Sandoz Clinical Assessment Geriatric (SCAG) in AD patients [254]. These were the only clinical trials of vinpocetine in AD that could be found at the time of this writing, and both seem to indicate that vinpocetine is ineffective for AD. By contrast, the evidence of vinpocetine for the treatment of all-cause dementia is inconclusive but relatively promising.
In a 2001 meta-analysis of 3 out of 39 studies that met inclusion criteria, 327 patients with dementia or cognitive impairment due to other disorders took vinpocetine or placebo [255]. The vinpocetine treatment group had significantly better scores on the Mini Mental Status Questionnaire (MMSQ), the SCAG, and the CGI. However, the authors noted that vinpocetine is not indicated for the treatment of AD due to the little and inconsistent data available for this indication [255].
In a 2003 Cochrane systematic review and meta-analysis of the three available unconfounded, double-blind, randomized, placebo-controlled clinical trials of vinpocetine for the treatment of 583 patients with dementia, 30 mg and 60 mg vinpocetine were found to produce significant benefit, including on the CGI and on the SKT attention and memory scale at up to and including 13 months. Due to the inconclusive nature of the data, however, the authors concluded vinpocetine is not indicated for dementia treatment [256].
In a 2012 clinical trial, moderately severe MCI patients who took vinpocetine for 18 months showed significant improvements in cognition on the MMSE and the Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-Cog), daily activity on the Activities of Daily Living, mood on the Hamilton Depression Scale, and overall change in disease status according to patients and investigators on the patient global impression of change and the clinical global impression of change [257].
Compared to placebo, vinpocetine 5 mg twice daily for 3 months in a 2014 pilot study resulted in significantly improved memory and concentration in 56 patients with cognitive impairment due to either dementia of various etiologies or epilepsy [258].
Therefore, the evidence about vinpocetine for AD is very limited, but the two trials available seem to show that vinpocetine is not effective for this indication. However, vinpocetinedoes appear to hold promise for MCI and dementia patients, although further clinical trials with larger patient cohorts are required to further evaluate its efficacy for these indications.
Nicergoline
The ergot derivative nicergoline inhibits Ca2+/calmodulin-dependent PDE1 and cGMP-stimulated PDE2 activity [259] but activates the high Km cAMP-degrading PDE from heart [260]. In addition, it non-competitively inhibits Ca2+/Mg2+-dependent brain adenosine triphosphatase (ATPase). It exerts a complex effect on brain Na+/K+ ATPase whereby it activates the Na+/K+ ATPase at low concentrations but inhibits it at high concentrations [260, 261]. It also acts as a potent α1A adrenergic receptor antagonist [262]. Nicergoline has been available since the 1970s in over 50 countries and has been used to treat a variety of conditions, including AD, vascular dementia, acute and chronic peripheral circulation disorders, and cerebral infarction [263, 264].
In preclinical studies, nicergoline has been shown to restore cognitive deficits in AD mice [265], upregulate BDNF [266], reverse the age-associated cholinergic deficit and enhance K+-induced acetylcholine release [267, 268], activate catalase [269], decrease oxidative stress [265, 269–271], inflammation [265, 266], andapoptosis [265, 272], and decrease levels of IL-1β, IL-6, and TNFα [266].
A 2001 Cochrane systematic review of 14 unconfounded, double-blind, randomized, placebo-controlled trials found that nicergoline provided mild to moderate dementia patients with significant improvements on the SCAG, the MMSE, and the CGI at 6 and 12 months [273]. According to this meta-analysis, there was no significant improvement on the ADAS-Cog at 6 or 12 months in a total of 342 patients, although there was a non-significant trend in favor of nicergoline on this scale [263, 273]. This is important since the ADAS-Cog is used exclusively with AD patients. The claim that differences on the ADAS-Cog were not significant at 6 months are based exclusively on Crook et al. 1996, an unpublished report from Pharmacia and Upjohn [273]. The claim that the change on the ADAS-Cog with nicergoline was not significant at 12 months is based on a 2001 multicenter, randomized, double-blind, placebo-controlled trial in 346 patients with probable mild to moderate AD, in which nicergoline 30 mg twice daily resulted in significantly improved cognition on the ADAS-Cog at 6 months and mean differences in ADAS-Cog scores between treated and control patients that grew increasingly large—including at 12 months [274]. According to the primary source poster abstract by Amaducci et al. 1999: “In the reduced patient cohort treated for 12 months, the ADAS-cog score difference between treatments after 3, 6, 9, 12 months was 0.89, 1.07, 1.45 and 1.64 respectively, increasingly in favour of nicergoline [274].” It was noted that AD patients receiving nicergoline improved over the course of the first 6 months and then deteriorated by month 12 [274]. However, the deterioration on the ADAS-Cog in nicergoline-treated AD patients was less severe than in placebo-treated patients, a benefit that became increasingly apparent up to and including at 12 months [274]. In other words, it appears that nicergoline resulted in a modest but significant improvement in AD symptoms on the ADAS-Cog at 6 monthsand produced no significant improvement at 12 months but rather slowed an increasingly precipitous decline at 12 months [274]. It is unclear why this study was included for the 12-month analysis but not for the 6-month analysis in the Cochrane review, nor is it clear whether this would change the conclusion of the Cochrane meta-analysis that the trend towards improvement on the ADAS-Cog at 6 months was not significant. The finding of significant improvement on the ADAS-Cog at 6 months was formally published after the Cochrane review article was in a journal in 2001 [275]. There was no mention in this formal paper of the trends toward a slowing of decline on the ADAS-Cog at 12 months [274, 275].
Two more recent trials, however, studied smaller groups of patientsand yielded negative results. In a 2017 uncontrolled pilot trial, 16 patients with early AD received 30 mg nicergoline twice daily for 1.5 years on average [276]. No statistically significant differences were noted in the severity of dementia, activities of daily living, cognition, and depressive symptoms between baseline and follow-up. However, nicergoline increased cerebral blood flow in frontal and parietal regions [276].A 2019 study in 22 patients with early AD found that, compared to acetylcholine esterase inhibitors alone, acetylcholine esterase inhibitors plus nicergoline preserved cerebral blood flow to the left temporal pole and the middle cingulate gyrus but did not result in any significant difference in dementia severity [277].
Nicergoline was found by a 2014 systematic review and meta-analysis to have a good safety profile, especially compared to other ergot derivatives, with no more adverse events reported than for placebo [278].
Therefore,it appears that nicergoline may be indicated for dementia patients [273]. There are mixed reports about whether the improvement on the ADAS-Cog at 6 months that nicergoline provided was significant, and there was a non-significant trend toward slower cognitive decline on this scale at 12 months [273–275]. However, two more recent pilot trials had disappointing results [276, 277]. This makes it inconclusive whether nicergoline is useful for the treatment of AD. Further clinical trials of nicergoline in AD involving more patients appear to be warranted.
Deprenyl/selegiline
Deprenyl, also known as selegiline, inhibits the calmodulin-dependent PDE1A2 and monoamine oxidase (MAO) [224, 279]. Discovered in the 1960s by Zoltan Ecseri at Hungarian drug company Chinoin, deprenyl is used clinically to treat Parkinson’s disease, major depressive disorder, and attention-deficit/hyperactivity disorder [280–282]. In preclinical studies, deprenyl has been found to increase CREB phosphorylation [283], activate Nrf2 [284–286] and catalase [283], decrease oxidative stress [284–287], inflammation [286], mitochondrial dysfunction [287], and apoptosis [287], and potentiate UVradiation-induced PARP1 activation yet downregulate PARP1 protein expression [288].
In the latest Cochrane systematic review and meta-analysis from 2003, which included 17 unconfounded, double-blind, randomized, placebo-controlled clinical trials, it was found selegiline significantly improved memory and cognition in AD patients at 4–6 weeks and 8–17 weeks, but not at 21–30, 35–56, or 65–82 weeks, and it improved activities of daily living at 4–6 weeks but not at later time points [289]. These 17 randomized controlled trials (RCTs) found significant improvements in memory on multiple scales, including the MMSE in 6 RCTs, the Randt Memory Index and the Wechsler Memory Scale in 2 RCTs each, and the Buschke Selective Reminding Test, prose recall, the Rey-AVL, and the ADAS-Cog in 1 RCT each. Sideeffects were reported, but very few were severe enough that a patient had to leave the trial, and there was no significant difference between the number of adverse events and trial withdrawals reported with selegiline and placebo. However, meta-analysis of the global rating scales (i.e., the Blessed Dementia Scale, Gottfries-Bråne-Steen scale (GBS), Global Deterioration Scale, and CGI) found no significant difference between selegiline and placebo. Birks and Flicker concluded that, “selegiline for Alzheimer’s disease has proved disappointing ... .there is [] no evidence of a clinically meaningful benefit for Alzheimer’s disease sufferers. This is true irrespective of the outcome measure evaluated, i.e. cognition, emotional state, activities of daily living, and global assessment, whether in the short, or longer term (up to 69 weeks), where this has been assessed [289].” Nevertheless, the authors’ own results do appear to point to modest and short-termyet statistically significant improvements in memory, cognition, and activities of daily living on various scales with selegiline treatment in AD patients, although the short-lived nature of these improvements do appear disappointing.
Cilostazol
PDE3 degrades cAMP and cGMP and is localized primarily to the endoplasmic reticulum membrane [290, 291]. Cilostazol is a PDE3 inhibitor used as an anti-platelet agent. Invented in the late 1980s by Otsuka Pharmaceutical Co., it is used clinically to treat intermittent claudication in peripheral vascular disease and to prevent stroke [292, 293]. Cilostazol has been shown in preclinical studies to rescue cognitive dysfunction and promote soluble Aβ brain efflux in a mouse model of cerebral amyloid angiopathy, decrease oxidative stress and apoptosis, suppress Aβ oligomerization and deposition [294, 295], inhibit GSK3β [168, 173], activate CREB [168, 295–299], induce mitochondrial biogenesis via cAMP/PKA/CREB/PGC1α signaling [298–301], induce Nrf2 and target gene expression [298, 302–305], decrease oxidative stress [306], upregulate SIRT1 protein levels [307], activate adenosine monophosphate-activated protein kinase [308, 309], inhibit NFκB [306–308, 310–312], downregulate BACE1 [307], and decrease levels of IL-1β [305], IL-6 [168, 305], and TNFα [298, 305].
Epidemiologically, taking cilostazol has been associated in a dose-dependent fashion with decreased risk of incident all-cause dementia in patients over 65 with an overall adjusted hazard ratio of 0.75 (meaning that patients over 65 taking cilostazol are 25% less likely to develop dementia compared to those not taking cilostazol), with high-dose cilostazol users having a 0.53 adjusted hazard ratio (meaning that high-dose cilostazol users are 47% less likely to develop dementia)[313]. Subgroup analysis revealed that cilostazol users who had ischemic heart disease or cerebral vascular diseases were significantly protected from dementia (adjusted hazard ratios 0.44 and 0.34, respectively) [313].
A retrospective study found that control patients deteriorated by ∼2 points on the MMSE per year, whereas cilostazol-treated patients had no change in MMSE scores per year, a simsignificant difference [314]. Subgroup analysis revealed that control MCI patients had an annual decrease in MMSE score of 4 points, whereas cilostazol-treated MCI patients had a very small, ∼0.2 point increase in MMSE score per year, a significant difference [314]. The differences between MMSE scores for healthy control patients and dementia patients were not significant, although it is worth mentioning that control dementia patients had an annual decrease in MMSE scores of ∼1.1, whereas cilostazol-treated dementia patients had an annual increase in MMSE scores of ∼0.4 points [314]. In other words, the cilostazol-treated dementia patients appeared to improve slightly more on the MMSE than cilostazol-treated MCI patients (though this trend was non-significant), but the effect of cilostazol in dementia patients may have been found to be non-significant simply because the control dementia patients deteriorated less rapidly than the control MCI patients did [314].
In another retrospective analysis comparing patients with mild dementia who received either donepezil alone or donepezil plus cilostazol, cilostazol users had a significantly slower rate of cognitive decline as measured by the MMSE [315].
In a 12-month case-control study in 60 patients with stable AD taking acetylcholine esterase inhibitors, cilostazol add-on usage was significantly associated with a decreased rate of cognitive decline on the MMSE [316].
In an open-label, uncontrolled pilot trial of 10 patients with AD, improvement in MMSE score was found at 6 months [69, 317]. In a randomized, controlled trial of 20 patients with AD and cerebrovascular disease, compared to taking aspirin or clopidogrel, taking 100 mg/day cilostazol was found to prevent decrements in scores on the Japanese ADAS-Cog, Trail Making Test-A, and the Revised Wechsler Memory Scale (logical memory-I) at 6 months. Only the control patients had decreased cerebral blood flow in the left middle temporal gyrus, whereas the cilostazol-treated patients had significantly increased blood flow in the right anterior cingulate lobe [318].
In a 6-month, randomized, double-blind, controlled trial of 36 patients with mild or moderate AD and white matter lesions, 100 mg cilostazol plus donepezil compared to donepezil alone prevented decreases in glucose metabolism in the parietal and frontal lobes compared to controls and preserved glucose metabolism in the left inferior frontal gyrus [319]. However, no significant differences were observed on the MMSE, the ADAS-Cog, or other outcome measures. That being said, cilostazol-induced improvements in glucose metabolism correlated with better scores on the ADAS-Cog [319]. Further trials of cilostazol are ongoing [69, 320].
Therefore, cilostazol usage may be associated with a decreased risk of dementia [313]. Itmay slow the rate of cognitive decline on the MMSE in MCI patients [314], mild dementia patients [315], and AD patients [69, 316–318]. However, the trial data about cilostazol in AD specifically is very limited and overall disappointing, with efficacy found by two pilot trials (one controlled, the other not) studying 30 total AD patients, but lack of efficacy found by a rigorous clinical trial studying 36 AD patients with white matter lesions [69, 317–319]. Based on these findings, future trials of cilostazol should focus on patients with MCI or early AD, especially those with comorbid cerebrovascular disease.
Denbufylline
Denbufylline inhibits PDE4, raising cAMP [321]. For unclear reasons, there is a dearth of preclinical evidence about denbufylline, although PDE4 inhibition is arguably one of the most promising PDE therapeutic targets for the purpose of cognitive enhancement and AD treatment according to the following preclinical evidence and a recent review of the preclinical evidence about PDE inhibitors for the treatment of AD [73]. For example, treatment with the PDE4 inhibitor rolipram resulted in long-lasting improvements in basal synaptic transmission, long-term potentiation, and working, reference, and associative memory in APP/PS1 transgenic AD mice [322]. In rats exposed to Aβ40 in the CA1 hippocampus, 0.5 mg/kg rolipram rescued memory deficits and CREB phosphorylation [323]. In rats microinfused with Aβ25–35 into the CA1 hippocampus, 0.1, 0.25 and 0.5 mg/kg/day intraperitoneal PDE4-inhibiting rolipram dose-dependently reversed memory deficits [324, 325], restored CREB phosphorylation and Bcl2 expression, decreased p65 NFκB and Bax expression [324], decreased ROS and malondialdehyde levels, rescued glutathione levels and SOD activity, upregulated thioredoxin, and inhibited the inducible iNOS pathway in the hippocampus [325]. In mice injected in the dentate gyrus with aggregated Aβ42, lentiviral RNA silencing of long-form PDE4D rescued Aβ42-induced cAMP decrements and memory deficits on the Morris water maze and the novel object recognition test while upregulating pSer133-CREB and BDNF and downregulating p65 NFκB, IL-1β, and TNFα [182]. In APPswe/PS1dE9 mice, 0.001 mg/kg of the PDE4D isoform-specific inhibitor GEBR-7b for three weeks at 5 months of age improved spatial memory on the object location test at 7 months of age without affecting CREB phosphorylation, BDNF expression, synaptic densities, Aβ levels, tau phosphorylation, or GSK3β activation [326]. Clinical trials have not been performed of any of these preclinically tested PDE4 inhibitors, possibly because the side-effect of emesis has hampered their clinical development [69]. However, these preclinical effects of PDE4 inhibition are likely reasonably representative of denbufylline’s preclinical effects.
In a randomized, multicenter, parallel group, double-blind, placebo-controlled clinical trial in 45 patients with mild to moderate AD not taking any other medications, 100 mg of denbufylline given twice daily for 3 months significantly improved scores on the SCAG, the CGI, the Digit Symbol Substitution Test, and the MMSE compared to placebo [327]. It also decreased delta activity and accelerated slow activity on imaging (which are increased in AD patients), indicating an increase in vigilance that correlates with symptomatic improvement [327]. Neither emesis nor any other adverse effects were reported [327].
In a trial in 226 AD patients and 110 patients with vascular or mixed dementia, 25, 50, or 100 mg denbufylline for 4 months significantly improved MMSE scores but only when these patients were considered as a single group [328]. Significantly more all-cause dementia patients who received denbufylline than those who received placebo had improved MMSE scores. No major adverse events were reported [328].
Therefore, the trial data about denbufylline in AD is very limited but somewhat promising. Emesis was not reported in association with this PDE4 inhibitor [327, 328]. Further trials of denbufylline in AD may be warranted.
Sildenafil
PDE5A specifically degrades cytoplasmic cGMP produced by sGC downstream of NO [71]. Sildenafil (also known as Viagra) inhibits PDE5, thereby raising cytoplasmic cGMP levels. Discovered in the late 1980s by Pfizer, sildenafil is used clinically to treat erectile dysfunction and pulmonary arterial hypertension [329, 330]. In preclinical rodent models of AD,sildenafil has been shown to rescue memory [177–180, 331–335], decrease Aβ levels [178, 333–336] and tau hyperphosphorylation [177, 331], inhibit GSK3β [177–180] and JNK [331], decrease IL-1β, IL-6, and TNF-α secretion [335], upregulate pSer133-CREB [180, 335], BDNF [180, 336], Arc [180], and Bcl2 [336], downregulate BACE1 [179], AβPP, caspase-3, and Bax [336], and decrease double-stranded DNA breaks and apoptotic cells [336]. For a recent review of sildenafil for AD, see Sanders 2020 [337].
Single-doses of 50 mg sildenafil have been shown in small groups of AD patients to decrease spontaneous neural activity in the right hippocampus, decrease cerebrovascular reactivity, and increase cerebral blood flow and the cerebral metabolic rate of oxygen [338, 339]. Based on this very limited preliminary evidence, clinical trials of sildenafil in AD patients are warranted.
A dose-limiting effect of sildenafil is that high doses of sildenafil raise cGMP levels to such an extent that cGMP-sensitive PDE2 is activated, degrading cAMP [72]. This effect has been shown to limit the effective dosage of sildenafil in metabolic conditions [72]. Therefore, future clinical trials should test sildenafil combined with a cGMP-stimulated PDE2 inhibitor, such as propentofylline [72, 340].
PF-04447943 and BI 409,306
In a phase II multicenter, double-blind, randomized, placebo-controlled trial, Pfizer’s PDE9A inhibitor PF-04447943 failed to improve scores significantly better than placebo on the ADAS-Cog, the Neuropsychiatric Inventory, or the CGI-Improvement scale [341].
In two multicenter, double-blind, parallel-group, randomized, placebo-controlled phase II studies, MCI and mild to moderate AD patients who took the PDE9 inhibitor BI 409306 developed by Boehringer Ingelheim did not show any significant difference on the Neuropsychological Test Battery total z-score at 12 weeks, nor on the ADAS-Cog11, the Clinical Dementia Rating scale-Sum of Boxes, or the Alzheimer’s Disease Cooperative Study-Activities of Daily Living scale [342].
In preclinical models of AD, PDE9 inhibition has been shown to rescue synaptic plasticity and memory and decrease dendritic spine density degeneration and cytotoxicity [213, 344]. However, PDE9 inhibitors PF-04447943 and BI 409306 have been shown to be ineffective in MCI and AD patients [341, 342]. This might bebecause PDE9A regulates natriuretic peptide-induced pGC-generated peri-plasma membrane cGMP, not NO/sGC-generated cytoplasmic cGMP [71]. As discussed, the NOS/NO/sGC pathway and cytoplasmic cGMP pools are disrupted in AD [195, 196], whereas NP/pGC/cGMP signaling and peri-plasma membrane cGMP are maintained or increased [195, 212–214], which might make increasing peri-plasma membrane cGMP with PDE9inhibitors a less viable therapeutic approach than replenishing cytoplasmic cGMP pools with PDE5 inhibitors [213].
Another consideration is that it is NP-activated pGC, not NO-activated sGC, that produces the peri-cytoplasmic cGMP that activates cyclic-nucleotide gated (CNG) channels [68]. Therefore, PDE9 inhibition might be expected to promote CNG activation, unlike PDE5 inhibition (Fig. 2) [68, 71]. CNG channels are non-specific cation channels that allow Ca2+ ions to influx into the cytoplasm of olfactory sensory neurons [345]. Excessive axonal Ca2+ influx induces Wallerian axonal degeneration, and excessive cytoplasmic Ca2+ influx induces excitotoxicity [346–349]. Olfactory sensory neurons are exposed to multiple sporadic AD risk factors, including combustion-derived carbon nanoparticles and microbial toxins [350–353]. AD pathogenic factors spread between neurons trans-synaptically in extracellular vesicles, such as exosomes [354–358], and tau pathology spreads through the brain in AD in a stereotypical fashion [2]. Olfactory sensory neurons’ axons project into the olfactory bulb [359], a brain region that is critically affected starting from the preclinical stages of AD [350, 351]. Thus, PDE9A inhibition might promote pGC/cGMP-activated CNG-mediated Ca2+ influx into olfactory sensory neurons, axon degeneration, excitotoxicity, and possibly propagation of AD pathologies to the olfactory bulb, possibly counterbalancing its positive effects and providing no net change in AD patients taking PDE9 inhibitors.
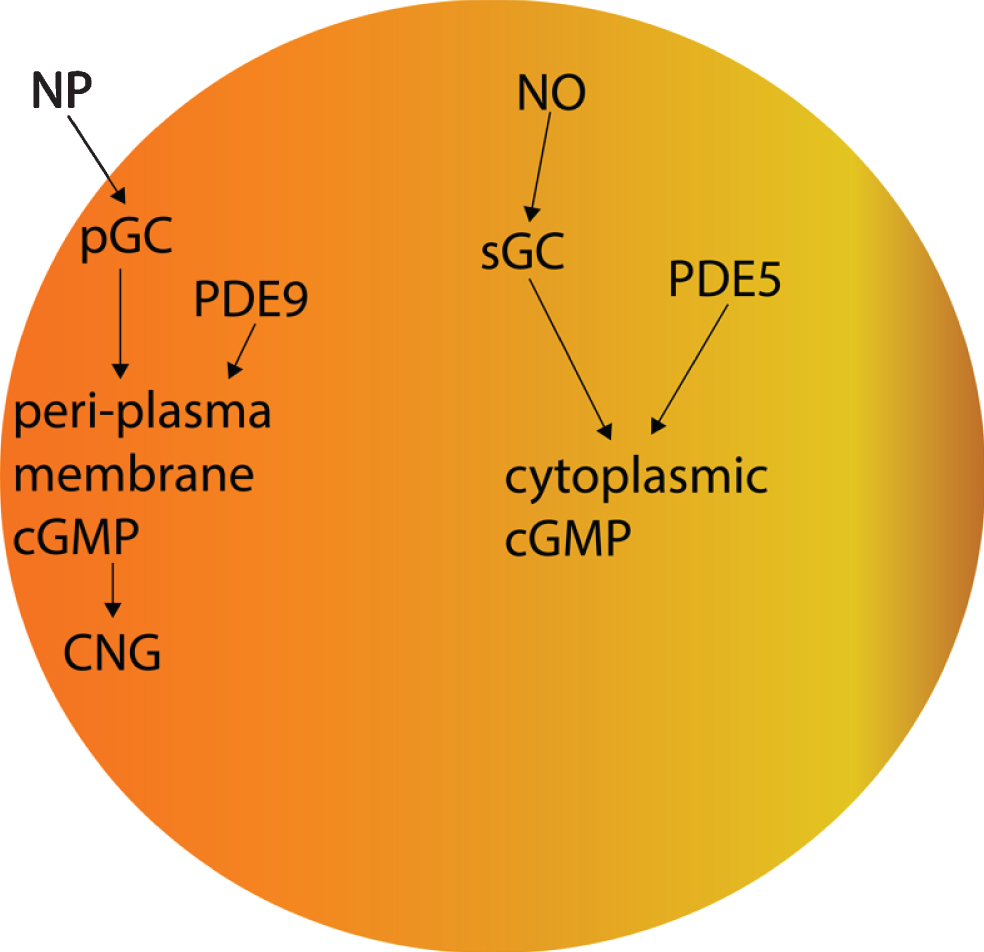
The effects of PDE5 versus those of PDE9 on cGMP.
Furthermore, it is unclear whether CNG channel expression is restricted to the AD brain olfactory sensory neurons, since the CNG subunit CNGA1 is significantly upregulated at the mRNA level in AD patients’ entorhinal cortices (Fig. 3) [360], and the cGMP-sensitive CNGA3 subunit is significantly upregulated in AD patients’ hippocampi (Fig. 4) [345, 360]. CNG mRNA was rarely expressed in a non-AD patients’ temporal cortex, but when it was, it was typically expressed by neurons (Figs. 5–10) [360, 361]. This suggests the possibility that PDE9 inhibitors might promote pGC/cGMP/CNG/Ca2+-mediated axonal degeneration and excitotoxicity in the sporadic AD entorhinal cortex and hippocampus.
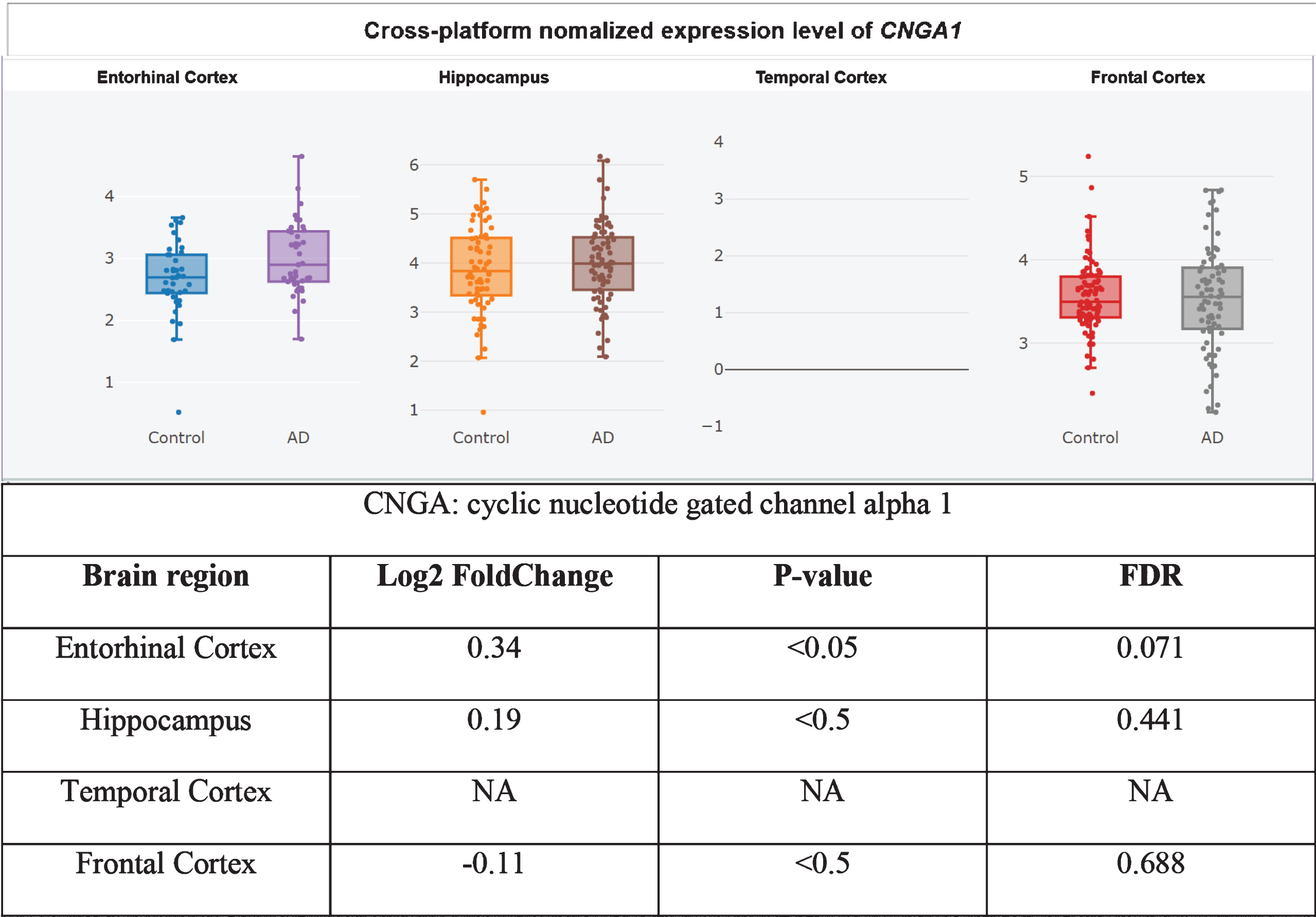
CNGA1 upregulated in AD entorhinal cortex, adapted from Xu et al. 2018 with permission [360].
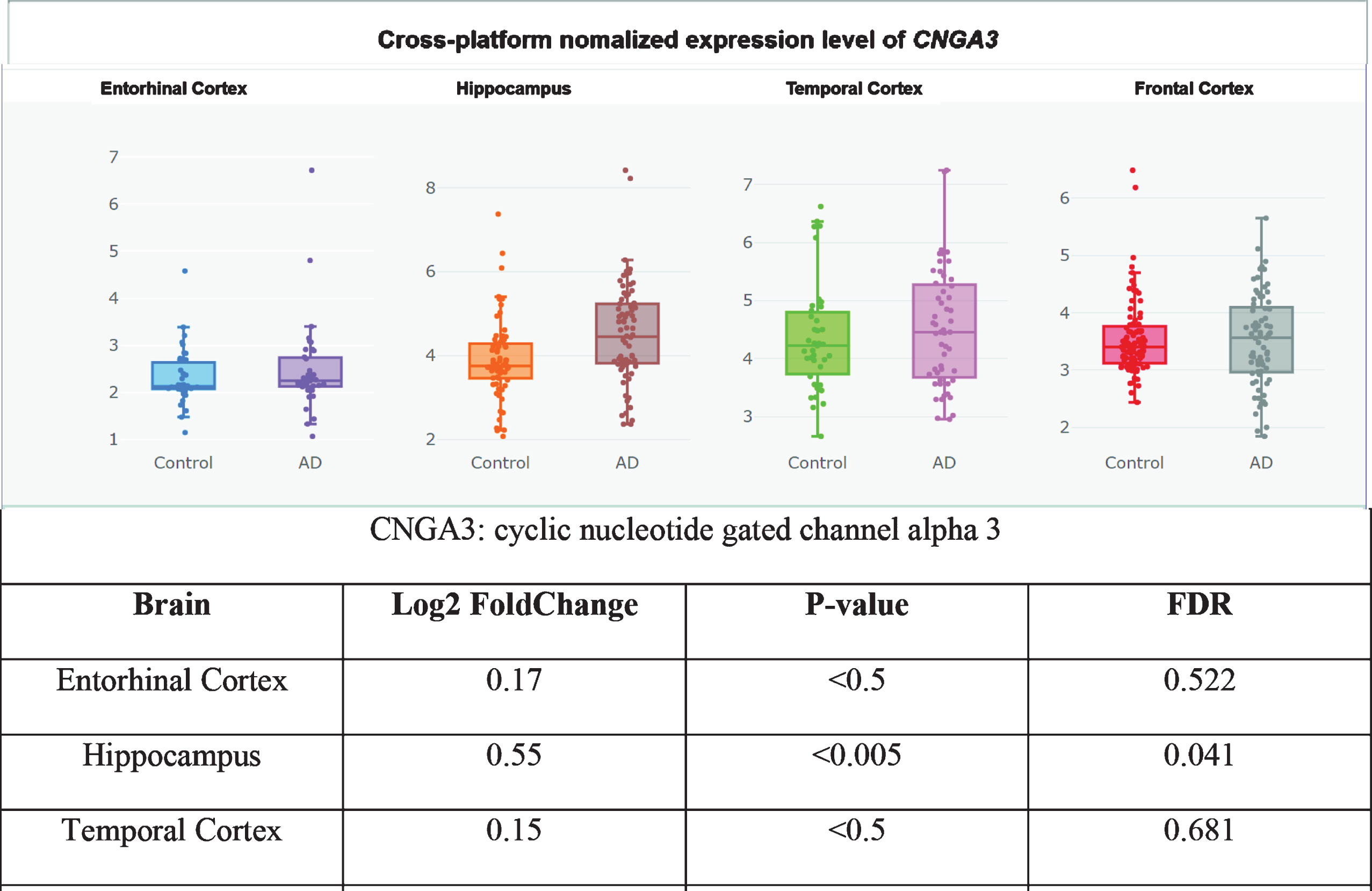
CNGA3 upregulated in AD hippocampus, adapted from Xu et al. 2018 with permission [360].
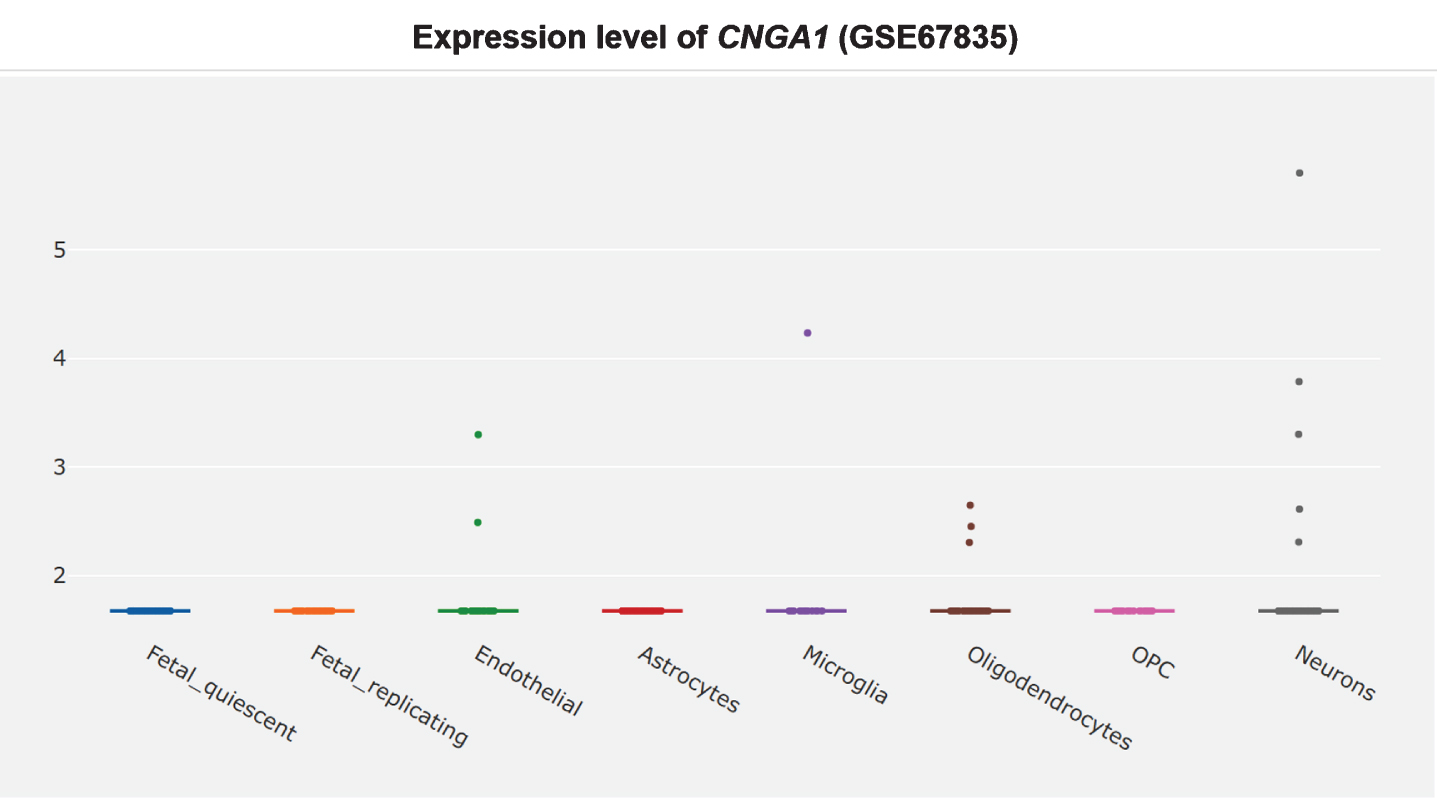
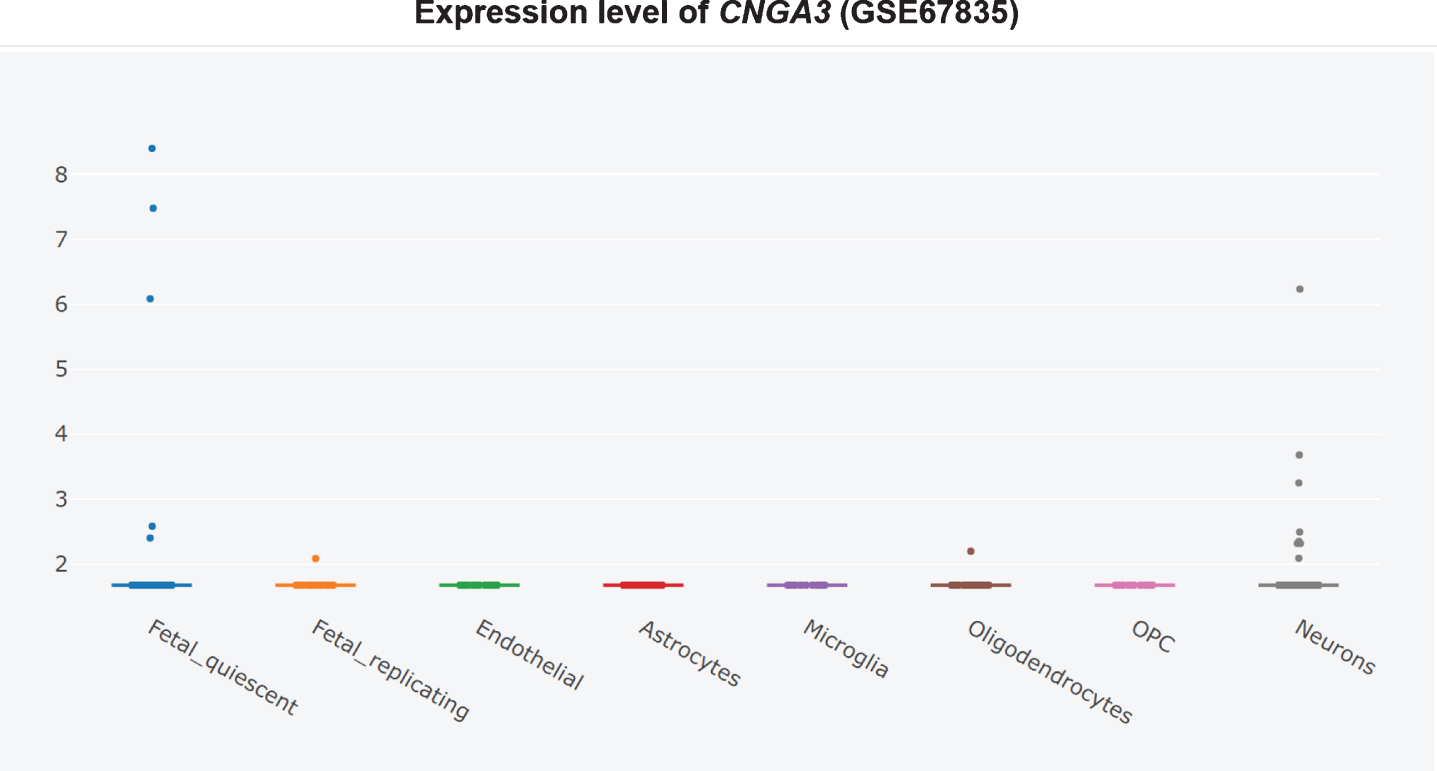

Therefore, PDE9 inhibitor might do as much harm as good, equaling out to no net effect in clinical trials. PDE9 inhibitors should not be pursued further for the treatment of AD.
Caffeine
Caffeine is a non-specific/broad-spectrum PDE inhibitor. Like propentofylline, caffeine is a methyl xanthine derivative [362, 363]. In AD transgenic mice, caffeine partially rescued PKA activity and CREB phosphorylation in the striatum but not the frontal cortex [364], decreased plasma, cortical, and hippocampal Aβ levels [365, 366], suppressed BACE1 and PS1 expression [366], and improved working memory [366].
The evidence about whether caffeine lowers the risk of AD, cognitive decline, or dementia is mixed. In a 2015 meta-analysis of 31,479 individuals enrolled in 20 studies, caffeine intake from coffee or tea was not significantly associated with the incidence of cognitive disorders [367]. In a 2016 study (n = 6,467), caffeine intake was associated with a lower risk of incident dementia and cognitive decline in women over the age of 65 [213, 368]. In a 2016 meta-analysis of 29,155 participants enrolled in 11 prospective studies, no association was found between caffeine intake and dementia or cognitive decline, but high caffeine intake was significantly associated with a decreased risk of incident AD [369]. In a 2017 meta-analysis of 34,282 individuals enrolled in 9 prospective cohort studies, a J-shaped association was found whereby 1–2 cups of coffee daily intake was associated with lower risk of cognitive disorders, but the association for 3 cups of coffee or more was not significant [370]. However, a 2018 dose-response meta-analysis of 8 prospective studies found no significant association between caffeine dosage and risk of AD or dementia [371]. Nevertheless, a 2019 study found that compared to drinking less than two cups of coffee a day, lifetime drinking two or more cups of coffee a day was significantly associated with reduced Aβ plaque burden in 411 cognitively-intact older adults [372]. Therefore, caffeine intake may be associated with a decreased risk of dementia, cognitive decline, and/or AD. No clinical trials of caffeine in AD have been conducted.
Propentofylline
Perhaps the most promising PDE inhibitor for the treatment of AD is propentofylline. Propentofylline is a methyl xanthine derivative like caffeine that acts as a relatively potent and nonspecific cAMP/cGMP PDE inhibitor and adenosine reuptake inhibitor [340, 363]. Propentofylline has been shown in preclinical models to suppress Aβ plaque deposition, tau hyperphosphorylation, GSK3β activation [373], microglial ROS generation [374–378], glutamate production, LPS-induced microglial IL-1β and TNFα secretion [375–377], Aβ-induced IL-1β secretion [373], and microglial proliferation [374–377]. It has been shown topromote anti-inflammatory regulatory T cell proliferation [379], restoration of ATP production [131, 132], and cerebral metabolic response to a memory task [380]. It can enhance irradiation-induced G1/S transition block [381]. It has also reversedaluminum-induced brain edema and hypoxia-like metabolic changes [382] and Aβ-induced memory deficits [383], as well as preventednerve cell death induced by either nerve growth factor withdrawal or Aβ [384–386].
Propentofylline has perhaps the most impressive history of phase III clinical trials of any of the PDE inhibitors reviewed in this manuscript.In a 1998 meta-analysis of four phase III, randomized, double-blind, placebo-controlled clinical trials ranging from 6 to 14 months, 300 mg of propentofylline taken thrice daily 1 hour before meals provided consistently significant improvements both in patients with AD and patients with vascular dementia [375]. These benefits persisted even after treatment cessation, suggesting a disease-modifying rather than a purely symptomatic improvement in these dementia patients [375]. Subgroup analysis revealed that, at 6 months, AD patients treated with propentofylline demonstrated significantly improved global function on the GBS, cognition on the MMSE and the SKT, and activities of daily living on the Nürnberger Altersbeobachtungs-skala (NAB questionnaire). At the final visit, treated AD patients exhibited significant improvements in all categories tested, including on the GBS, CGI item II (global improvement), SKT, MMSE, and NAB [375].
A 2003 Cochrane systematic review of unconfounded double-blind, randomized, placebo-controlled clinical trials in dementia patients found that propentofylline treatment resulted insignificantly improved cognition at 3, 6, and 12 months (including on the MMSE at 12 months), global assessment at 3 months, dementia severity at 3, 6, and 12 months (including on the CGI at 12 months), and activities of daily living at 6 and 12 months [376]. However, data from an additional 1200 patients in randomized clinical trials was not shared with the authors by the drug’s manufacturer, Aventis [376]. An update on this systematic literature search performed in 2008 yielded no new clinical trials of propentofylline in dementia patients [376].
In a 1999 18-month-long multinational, randomized, double-blind, placebo-controlled clinical trial in 486 mild to moderate AD patients, 300 mg propentofylline thrice daily 1 hour before meals resulted in significantly improved cognitive performance on the ADAS-Cog and global function on the Clinician’s Interview-Based Impression of Change Plus Caregiver Input (CIBIC-Plus) [376, 387–391]. Benefits persisted 6 months after treatment cessation in the 18-month clinical trial as well, further suggesting a disease-modifying effect [387, 391]. The results of this trial were presented in several poster abstracts [376, 387–391]. This study was listed in the Cochrane review but not included in the meta-analysis due to insufficient raw data from it being available [376].
However, a 2007 book chapter claimed that after two successful phase III clinical trials (there were more than two), an18-month trial “failed to show benefit, and development was discontinued [392].” The author did not cite this claim, making its source unclear. However, a MedScape article published in 2000claimed that the 18-month-long Propentofylline Long-term Use Study (PLUS) showed no significant difference between treatment and placebo in AD patients, leading Aventis to discontinue development of the drug [393]. According to this article, in the late 1990s, the European Agency for the Evaluation of Medicinal Products rejected Aventis’s application to market propentofylline for the treatment of AD and vascular dementia [393], andin 1999, the manufacturer announced a phase III trial of the drug in AD patients to address “regulatory concerns,” but this was later cancelled [393]. The article does not contain any references. A 2008 book reproduced the preceding claimswithout citing its source [394]. Neither of these books nor the MedScape article reference the successful 18-month-long phase III clinical trial presented in the 1999 poster abstracts. It is unclear to this author whether there were two 18-month trials, one successful and one not, or one single trial that this MedScape article and the subsequent two books misreported. Regarding safety, the drug was well tolerated in these studies for periods of up to 18 months [376, 395].
Now, in the UK, propentofylline is used for the treatment of canine cognitive dysfunction [396], which has been proposed as a superior preclinical model of AD compared to rodent models since dogs naturally develop age-associated Aβ plaques with AD-like symptoms [397].
An economic analysis found that adding propentofylline to the standard of care for patients with mild to moderate AD in Sweden would save 3.8–7.6% of the costs associated with caring for this population [398]. A Canadian economic analysis based on clinical trials of 12 months’ duration found that treating dementia patients with propentofylline would improve health outcomes and reduce home care and caregiver costs [399].
Propentofylline is the most effective inhibitor out of four PDE-inhibiting xanthine derivates tested (i.e., pentoxifylline, propentofylline, torbafylline, and albifylline) for all of the PDE isoforms that were measured (i.e., PDE1, PDE2, PDE3, and PDE4), with it being particularly effective at inhibiting cGMP-stimulated PDE2 and PDE4 [340]. This is a remarkable selectivity profile for several reasons.First, this makes propentofylline the only PDE inhibitor reviewed here that inhibits more than one of the most promising to inhibit in AD PDEs (i.e., PDE2 and PDE4) [69, 340]. Second, inhibiting multiple PDEs and boosting both cAMP and cGMP might offer synergistic improvement of CREB, PGC1α, and Nrf2, etc., signaling [72]. Third, high doses of PDE5 inhibitor sildenafil have the side-effect of activating cGMP-sensitive PDE2 [72], resulting in cAMP degradation and potentially disrupting PKA-mediated CREB and PGC1α signaling [72, 93]. This suggests that propentofylline and sildenafil might provide AD and vascular dementia patients with synergistic disease-modifying benefit by inhibiting PDE2, PDE4, and PDE5 simultaneously. Propentofylline, sildenafil, donepezil, and memantine should be compared to donepezil and memantine (standard of care) alone in future preclinical studies and clinical trials for the treatment of AD.
CONCLUSION
Based on the preceding discussion, it can be concluded that modulating cyclic purine nucleotide levels with certain PDE inhibitors can prevent and treat AD, MCI, and dementia in a disease-modifying fashion (see Table 1 for summary of the clinical effectiveness of the PDE inhibitors discussed). Caffeine and cilostazol may be associated with a decreased risk of incident dementia, cognitive decline, and AD [213, 372], suggesting that PDE inhibitors may help prevent AD. The clinical trials of denbufylline and sildenafil are promising but very preliminary [327, 339], so no conclusions can be drawn from them. Clinical trials of PF-04447943 and BI 409,306 have shown a lack of efficacy [341, 342], which might be because PDE9 inhibition increases peri-plasma membrane cGMP (rather than cytoplasmic cGMP) and allows Ca2+ influx through CNG [68, 213]. The clinical trials of vinpocetine [253, 255–258], cilostazol [69, 317–319], and nicergoline are preliminary and mixed [273–277], with cilostazol and nicergoline likely being the most promising and the worthiest of further study of these three [273–277]. Deprenyl/selegiline clinical trials show short-term but not long-term benefits, making deprenyl somewhat disappointing [289]. By contrast, propentofylline has been shown in five phase III clinical trials to improve cognition, dementia severity, activities of daily living, and global assessment in mild-to-moderate AD patients on multiple scales [375, 387–391], including on the ADAS-Cog and the CIBIC-Plus at 18month in a phase III clinical trial that was presented in several 1999 poster abstracts [376, 387–391]. However, two books published in 2007 and 2008, respectively, claimed based, apparently, on a MedScape article published in 2000 that an 18-month phase IIIb clinical trial failed to show efficacy, so propentofylline was discontinued [392–394]. It is unclear whether there were two 18-month-long phase III trials of propentofylline, or whether the MedScape article was inaccurate. Ultimately, propentofylline may be indicated for the treatment of patients with mild-to-moderate sporadic AD. Regardless, propentofylline is used now to treat canine cognitive dysfunction [396]. Like human AD, canine cognitive dysfunction involves age-associated wild-type Aβ deposition, making it a superior preclinical model of AD compared to rodent models [397]. Ironically, this impliesthe hypothesis that if propentofylline treats canine cognitive dysfunction, then it may treat human sporadic AD.
Treatment effects of propentofylline persisted months after treatment cessation in multiple trials, suggesting a disease-modifying effect in AD patients [375, 391]. We propose this disease-modifying effect may have been observed because, as a potent and non-specific/broad-spectrum PDE inhibitor [340], propentofylline raises cytoplasmic cAMP and cGMPenough to substantially enhance neural pSer133-CREB, SIRT1, PGC1α, and Nrf2 signaling [74–81, 144], improving neural synaptogenesis [84–90], memory [84], mitochondrial biogenesis [83, 103], antioxidant [112], detoxification, and survival gene expression [84–90] while also inhibiting BACE1-inducing inflammatory NFκB [34, 156–158] and tau-phosphorylating and CREB-inhibiting GSK3β [159–162, 177–181].
Interestingly, the only PDE inhibitor that has completed efficacy-testing clinical trials as of 2020 that is known to inhibit more than one of the most promising PDEs to inhibit to treat AD according a review of preclinical studies (i.e., PDE2, PDE4, and PDE5) [73] is propentofylline [340]. Propentofylline is known to inhibit PDE2 and PDE4 [340], two of the three most promising therapeutic targets [73]. It is perhaps unsurprising, then, that propentofylline is also the PDE inhibitor with arguably the most promising clinical trials so far [375, 387–391]. However, it is unknown whether propentofylline inhibits PDE5 as well [340], although it is a non-specific/broad-spectrum PDE inhibitor that raises both cAMP and cGMP, so it might [340]. If it does not, then the PDE5 inhibitor sildenafil might be the ideal therapeutic to co-administer with propentofylline. Furthermore, by potently inhibiting cGMP-stimulated PDE2 [340], propentofylline would be ideal to co-administer with sildenafil to prevent the dose-limiting side-effect of sildenafil of activating cGMP-stimulated PDE2 [72]. Future preclinical studies should study the effect of propentofylline and sildenafil together and apart plus standard of care compared to standard of care alone in AD to determine whether propentofylline and sildenafil combination treatment might provide synergistic benefits. Future clinical trials should investigate whether propentofylline improves AD symptom severity on the ADAS-Cog and overall impression in a third (or second?) 18-month phase III clinical trial in mild-to-moderate AD patients.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.