
Abstract
Lutzomyia longipalpis, is the main vector of visceral leishmaniasis in the Neotropic and the taxonomic identification is relevant for epidemiologic studies and for the disease control. The evidence supporting the existence of a L. longipalpis species complex morphologically indistinguishable besides similarity with females belonging the Lutzomyia subgenus and longipalpis series, difficult taxonomical identification when there are not available males. In this study, we used the sequence proposed worldwide as DNA barcode to distinguish animal species to characterize specimens belonging to L. longipalpis from three Colombian localities (Neiva, El Callejón, Girón) and evaluate their usefulness in separating them from closely related species such as L. gomezi, L. cruciata and L. bifoliata. The amplified and sequenced fragment exhibited a length of 548 bp and 26 haplotypes for 33 individuals of L. longipalpis, one haplotype for L.gomezi, one haplotype for L. cruciata and two haplotypes for L. bifoliata were found. Genetic distances (K2P) between L. longipalpis haplotypes (0.05-0.07) and clusters in a NJ dendrogram, effectively separated L. longipalpis from individuals belonging to closely related species. Individuals of L. longipalpis were separated in two groups, one included haplotypes from Neiva and El Callejón, and the other from Girón. The genetic distance found between the two groups of L. longipalpis were significantly higher than those found at the intraspecific level for species previously studied on the basis of the barcode sequence as L. trinidadensis (0.042) and L.panamensis (0.02).
Introducción
Lutzomyia longipalpis (Lutz & Neiva, 1912) (Diptera: Psychodidae), es una especie del subgénero Lutzomyia, caracterizada por la presencia de una corta espermateca anillada y un par de setas simples y persistentes en la base de la coxita del macho, también, en el parámero, se encuentra un par de setas curvadas en el margen dorsal medio (Young y Duncan 1994). Esta especie se considera el principal vector de leishmaniasis visceral en el Neotrópico y fue descrita por primera vez a partir de colecciones hechas en Brasil por Lutz y Neiva en 1912 (Grimaldi et al. 1989; Lainson y Rangel 2005).
La distribución geográfica de L. longipalpis va desde el Sur de México hasta el norte de Argentina, pero es discontinua, ocurriendo primariamente en zonas con hábitats secos en Centroamérica y el norte de Suramérica, aunque también ha sido asociada con bosque húmedo en la bahía ribereña del Amazonas (Lanzaro et al.1993).
En Colombia, L. longipalpis se encuentra asociado a bosque seco tropical de áreas del valle del río Magdalena, los departamentos de Santander y localidades de la costa caribe colombiana como Córdoba, Sucre y Guajira (González et al. 2006); sin embargo, en años recientes ha sido también colectada aunque en muy bajas densidades, en localidades y ecosistemas donde no había sido previamente registrada, ampliándo su rango geográfico de distribución (Travi et al. 2002; Flórez et al. 2006; Viveros et al. 2010).
Diversos estudios han señalado un considerable aislamiento geográfico entre las poblaciones de L. longipalpis relacionado con su baja capacidad de vuelo, poca dispersión, y presencia de barreras climáticas y geográficas entre los sitios donde se encuentra (Morrison et al.1993; Arrivillaga et al. 2002). La alta divergencia genética entre poblaciones y otras diferencias como las encontradas a nivel molecular en los péptidos producidos por la saliva (Maxadilan) de individuos de regiones geográficas diferentes (Warburg et al. 1994; Lanzaro et al. 1999; Yin et al. 2000), han sugerido la existencia de un complejo de especies, las cuales podrían a su vez diferir en aspectos relacionados con su papel como vector (Ward et al.1983; Ward et al. 1985; Lanzaro et al. 1993; Mutebi et al. 1998; Yin et al. 1999; Uribe 1999; Uribe et al. 2001; Arrivillaga et al. 2002, 2003; Watts et al. 2005). Adicionalmente, las hembras de L. longipalpis presentan rasgos morfológicos en las espermatecas muy similares a las del subgénero Lutzomyia y la serie longipalpis, y en especial a las de la especie Lutzomyia cruzi Mangabeira, 1938 (Martins et al. 1984; Young y Duncan 1994; Lanzaro y Warburg 1995; Vigoder et al. 2010). Por lo general, las características morfológicas de las espermatecas son fundamentales en la identificación taxonómica y en ausencia de machos, se dificulta la identificación y separación de las especies en zonas donde ocurren de forma simpátrica. En este contexto, disponer de una herramienta como las secuencias del código de barras genético para diferenciar L. longipalpis de forma rápida y eficiente sería de gran utilidad.
La iniciativa código de barras (BOLD) asigna secuencias de un fragmento del gen mitocondrial Citocromo oxidasa I (COI) a especies animales para facilitar el inventario de biodiversidad y la identificación de especies (Hebert et al. 2003), y aparece como una excelente herramienta en el caso de insectos de importancia médica, donde se requiere saber de forma rápida y acertada cuales son las especies presentes en un área de transmisión (Besansky et al. 2003; Azpurua et al. 2010; Jinbo et al. 2011).
La iniciativa ha tenido gran acogida por la conectividad y el lenguaje común de las secuencias de ADN, que permite a los investigadores de diferentes partes del mundo avanzar en estudios de taxonomía y sistemática de diversos grupos de organismos y en este caso particular de insectos vectores de enfermedades. Sin embargo, problemas como secuencias nucleares de origen mitocondrial (NUMTs), endosimbiontes, utilidad relativa en filogenia, concepto de especie, estandarización metodológica en el grupo de estudio, y la controversia en el uso de un solo marcador para un amplio rango de taxa (Moritz y Cicero 2004; Meier et al. 2006; Song et al. 2008; Casiraghi et al. 2010) hacen indispensables evaluar el fragmento propuesto como código de barras en cada grupo de especies a investigar, para validar su utilidad particular.
Es claro, que la iniciativa código de barras no es funcional para todos los grupos de especies y la variabilidad que exhibe, permite niveles de resolución que varían a lo largo de los grupos taxonómicos (Meier et al. 2006).
La tipificación molecular con haplotipos de la secuencia barcode a ejemplares del género Lutzomyia se ha iniciado recientemente incluyendo ejemplares de Panamá (Azpurua et al. 2010), con la idea de su uso potencial para diferenciar especies de importancia en la transmisión de leishmaniasis. En este sentido, el presente estudio pone a disposición de la comunidad científica las secuencias del principal vector de esta enfermedad (forma visceral) en Colombia y en particular de especímenes provenientes de los sitios donde se han registrado los importantes brotes de transmisión. Se asignan haplotipos de COI a especimenes machos L. longipalpis provenientes de las localidades de Neiva, El Callejón y Girón (Colombia).
Secuencias "barcode" de esta especie no se encontraron disponibles en las bases de datos de Genbank o CBOL al momento de la realización del estudio.
Materiales y Métodos
Los sitios de muestreo para L. longipalpis en Colombia son representativos de las regiones y ecosistemas colombianos donde se encuentra esta especie, de acuerdo con registros previos (Lanzaro et al. 1998; Uribe et al. 2001) y que fueron de fácil acceso en términos de seguridad de los investigadores. Los sitios seleccionados fueron: El Callejón (Departamento - Cundinamarca) (4°17′00,85″N 74°02′01,71"W), Girón (Departamento - Santander) (6°59′33,33"N -73°03′03″ W) y Neiva (Departamento - Huila) (2°54′53,28″N 75°16′46,26″W), las cuales son áreas activas para la transmisión de leishmaniasis visceral (González et al. 2006). Las capturas se realizaron entre los años 2009 y 2010, con ocho trampas de luz tipo CDC miniatura incluyendo un área lineal de 500 metros desde el domicilio hasta relictos de bosque. Las trampas se dejaron funcionando desde las 18:00 p.m hasta las 6:00 a.m del día siguiente. Adicionalmente se usó trampa Shannon en inmediaciones del domicilio, desde las 18:00 a las 24:00 p.m.
Los individuos se almacenaron en viales individuales EppendorfⓇ de 1,5 ml y se llevaron al laboratorio para su posterior procesamiento e identificación. Bajo estereomicroscopio y en una gota de solución salina, los especímenes fueron procesados así: se cortó la cabeza, alas, y abdomen, que posteriormente fueron aclarados en lacto-fenol (1:1) para la identificación morfológica. El tórax y las patas se removieron cuidadosamente para la extracción de ADN, manteniéndose a 4°C en una gota de isopropanol al 100%. Para la observación morfológica detallada y verificación taxonómica tradicional, las partes correspondientes se montaron en lámina porta y cubreobjetos con bálsamo de Canadá. Para la determinación taxonómica se usaron las claves de Young (1979), Young y Duncan (1994), y Galati (2009) y se contó con la ayuda del experto Charles Porter del CDC (Center For Disease Control, Atlanta).
Igual procedimiento se realizó para los especímenes de las especies Lutzomyia bifoliata (Osorno, Morales, Osorno & Hoyos, 1970), Lutzomyia gomezi (Nitzulescu, 1931) (colectadas en Río Claro-Antioquia, Colombia) y Lutzomyia cruciata (Coquillett, 1907) (San Francisco del Coray - Honduras) que fueron incluidos en el estudio como punto de comparación en términos de distancias y agrupaciones.
Para la extracción de ADN se uso el método Porter y Collins (1991), y las partes removidas de los individuos confirmados taxonómicamente como L. longipalpis. Los oligonucleótidos que se utilizaron para amplificar el fragmento "código de barras" de Citocromo Oxidasa I (548 pb), fueron LCO1490 GGTCAACAAATCATAAAGATATTGG (forward) y HCO2198 TAAACTTCAGGGTGACCAAAAAATCA (reverse) (Hebert et al. 2003).
La reacción en cadena de la polimerasa (PCR) fue llevada a cabo en un termociclador PTC-100 (MJ Research) bajo las siguientes condiciones: una desnaturalización inicial a 94°C por 5 minutos; seguido de 35 ciclos a 94°C por un minuto, 45°C a 1:50 minutos, 72°C a 1:50 minutos, y un paso final de 72°C a 5 minutos. La PCR se realizó en un volumen de 50 uL que contenía: buffer PCR 10X (NH4SO4), MgCl2 (25 mM), oligonucleótidos (2 mM), DNTPs (100 mM), 4 uL ADNmuestra y 0,5 unidades de Taq polimerasa (Fermentas®).
Para visualizar los productos de la PCR se preparó un gel de agarosa al 1% en buffer TBE 1X (Tris-Borato 40 mM, EDTA 1 mM a pH = 8.0), para sembrar 6 uL de cada muestra-producto de PCR (4/5 de ADN problema y 1/5 de EZ-Vission) con los controles positivos y negativos, y marcador de peso molecular (DNA Ladder-100 pb). Al gel se le aplicaron un voltaje de 90 voltios con una fuente de poder, durante 45 minutos para que el ADN migrara y luego se transfirió a una fuente de luz U.V para observar el resultado y fotografiar con cámara digital (Easy Kodak Share).
El fragmento amplificado se secuenció en ambos sentidos de la cadena, en un secuenciador automático ABI 310 en el Center of Disease Control (CDC - USA) gracias a todos los trabajos colaborativos con esa entidad. Los cromatogramas fueron editados manualmente en Bioedit v7.0.9 (Hall 1999) у alineados en ClustalW (Larkin et al. 2007) teniendo en cuenta la secuencia de referencia para Citocromo oxidasa I de Aedes aegypti (Linnaeus, 1762) (Genbank: NC_010241.1), y posteriormente evaluar la composición nucleotídica, variabilidad nucleotídica por sitio (entropía) y caracterización de mutaciones no informativas (patrón de saturación) mediante el software DAMBE (Xia 2002).
La descripción de la variabilidad de L. longipalpis y su comparación con los especímenes de las otras especies cercanas se realizó en términos de la variabilidad haplotípica, divergencias en las distancias genéticas correspondientes y sustituciones entre las secuencias.
El alineamiento final fue utilizado para calcular distancias métricas usando el modelo biparamétrico de Kimura (K2P) (Nei y Kumar 2000). Se calcularon diferencias nucleotídicas pareadas entre secuencias para haplotipos por localidad de L. longipalpis y secuencias de las especies relacionadas para calcular patrones de divergencia intra/interespecífica, además se realizó un dendrograma de Neighbor-Joining para representar las distancias genéticas mediante el software MEGA v4.1 (Tamura et al. 2007), teniendo en cuenta una prueba de bootstrap (1000 réplicas) para soportar las agrupaciones inferidas. También se utilizaron las secuencias registradas previamente por Azpurua et al. (2010), para construir un dendrograma de Neighbor-Joining (bootstrap = 1000 réplicas) y registrar la formación de agrupamientos y separación de especies del género Lutzomyia y los haplotipos colombianos de L. longipalpis secuenciados y alineados.
Resultados
El alineamiento fue de 548 pb y correspondió a las posiciones 1433-1947 del gen Citocromo Oxidasa I de A. aegypti, este segmento corresponde al propuesto por Hebert et al. (2003) como código de barras para la identificación de especies. Las secuencias mostraron un alto sesgo de A + T (X = 68%) en relación con el contenido G + C (X = 32%), como se ha registrado en los genes mitocondriales de artrópodos e insectos (Crease 1999; Hoy 2006). El contenido individual promedio de nucleótidos fue: A = 39,3%, G = 15,2%, C = 16,8%, T = 28,7%.
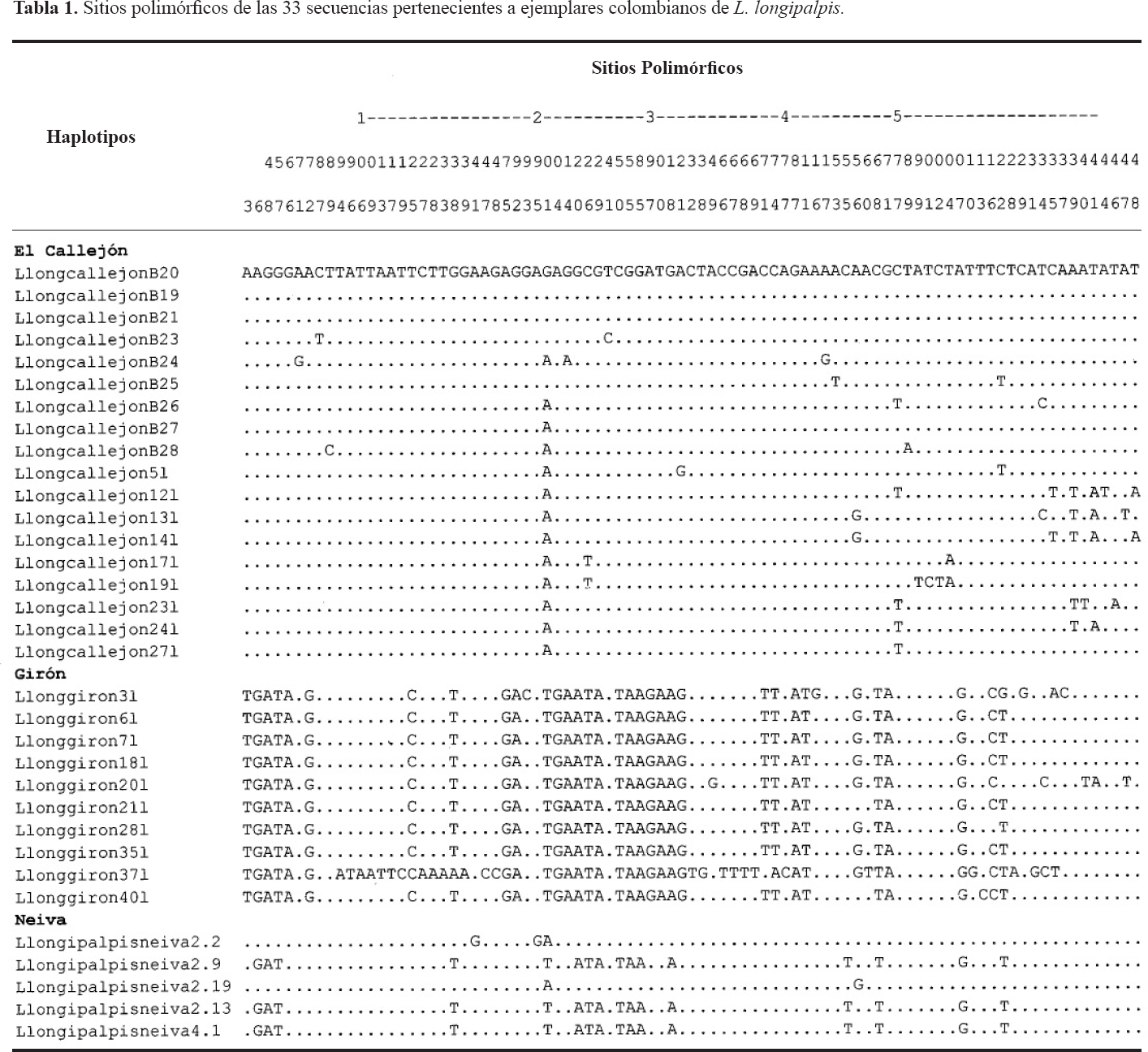
El alineamiento permitió discriminar 461 sitios conservados y 87 sitios variables (41 sitios parsimoniosamente informativos) (Tabla 1): 37 cambios sinónimos en la tercera posición del codón, 49 no sinónimas y un sitio que no pudo ser categorizado ya que hacía falta un nucleótido para completar el último codón. Se caracterizaron 26 haplotipos en las 33 secuencias y en la mayoría de las localidades se obtuvieron únicos haplotipos a excepción de cuatro individuos de Girón (LlongipalpisGiron6l, LlongipalpisGiron71, LlongipalpisGiron181, LlongipalpisGiron351), tres individuos de El Callejón (LlongipalpisElCallejonB19, LlongipalpisElCallejonB20, LlongipalpisElCallejonB21) y tres individuos de Neiva (LlongipalpisNeiva2.9, LlongipalpisNeiva2.13, LlongipalpisNeiva4.1); El mayor número de haplotipos fue observado en Callejón y Girón.
Sitios polimórficos de las 33 secuencias pertenecientes a ejemplares colombianos de L. longipalpis.
El análisis de entropía evidenció una variabilidad constante a lo largo del fragmento con tendencia a aumentar en las posiciones 190-255 (12 sustituciones) y 500-548 (21 sustituciones) (Fig. 1). La relación entre transiciones/transversiones, mostró un aumento gradual de las tasas de sustitución nucleotídica con respecto a las distancias génicas entre las especies, especialmente de las transiciones, aunque sin presentar saturación de sustituciones (Fig. 2).
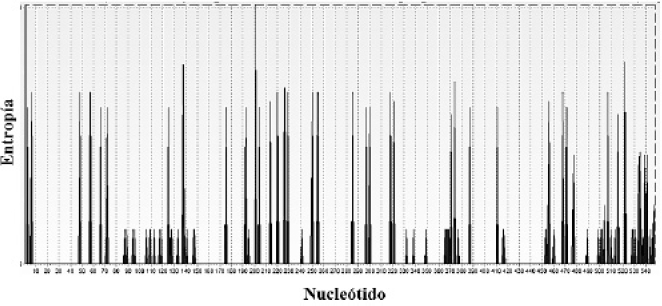
Entropía de la secuencia de nucleótidos dentro de la región del gen Citocromo oxidasa I - código de barras.
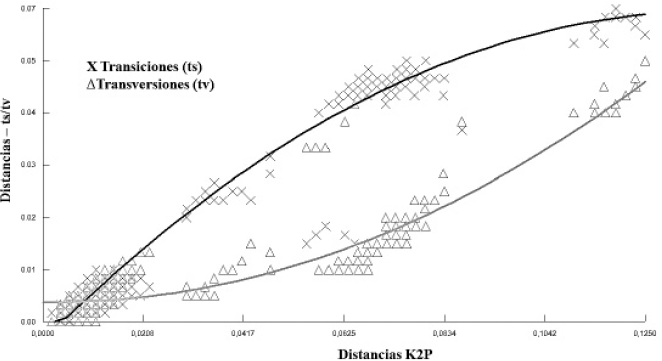
Distancias genéticas estimadas con el modelo Kimura-2 parámetros en relación con las tasas de transiciones y transversiones.
Las distancias genéticas (K2P) calculadas para haplotipos de L. longipalpis por localidad fueron variables presentando un intervalo de 5,0% (Neiva) a 7,0% (Girón), estos valores intra-específicos son mayores a los registrados para otras especies de amplia distribución e importancia vectorial (L. trinidadensis, L.gomezi, L. panamensis: < 4,0%) (Azpurua et al. 2010), sin embargo, las distancias inter-especificas con L. bifoliata, L. gomezi y L. cruciata se ubicaron entre el 12-18% originando una brecha con los haplotipos de L. longipalpis pertenecientes a diferentes localidades (Tabla 2). El dendrograma de Neighbor-Joining (Fig. 3) permitió agrupar y diferenciar los haplotipos de L. longipalpis de otras especies del subgénero Lutzomyia con significativos valores de bootstrapp (> 70), mostrando la utilidad del fragmento secuenciado para distinguir a L. longipalpis de especies del subgénero Lutzomyia cercanas evolutivamente. A nivel intraespecifico a pesar de las politomías entre haplotipos de una misma localidad, es evidente la formación de agrupamientos discretos entre secuencias locales, con excepción de dos haplotipos de Neiva (L.longNeiva2.2 y L.longNeiva2.19) que se ubican dentro del grupo formado por los haplotipos de El Callejón. El dendrograma construido utilizando las secuencias caracterizadas por Azpurua et al. (2010) en Genbank, permitió separar claramente a L. longipalpis de otras especies pertenecientes a diferentes subgéneros, grupos y series del género Lutzomyia (Fig. 4).
Valores de distancias genéticas bajo el modelo biparamétrico de Kimura. Las distancias genéticas estimadas se muestran en negrillas y en itálica la desviación estándar.
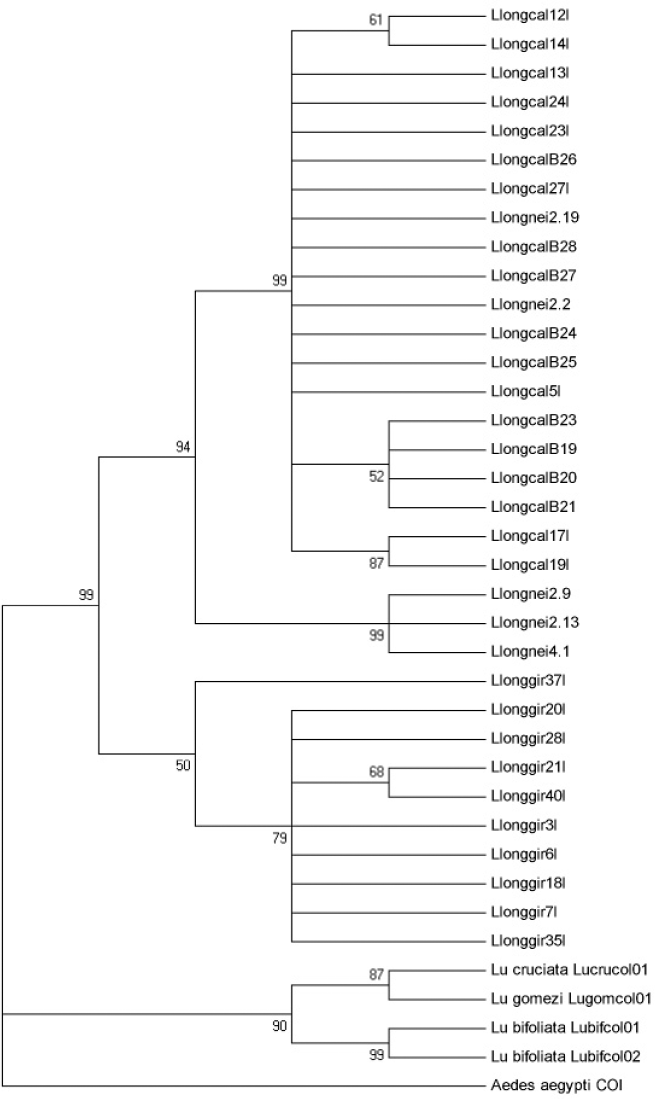
Dendrograma de Neighbor-Joining con el modelo K2P y bootstrapp de 1000 réplicas, mostrando valores de bootstrap cuando estos son superiores al 50% en los haplotipos colombianos del fragmento COI-barcode para L. longipalpis.
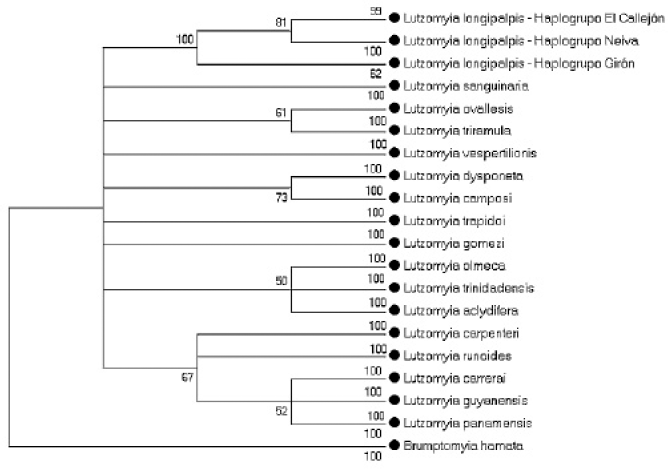
Dendrograma de Neighbor-Joining con el modelo K2P y bootstrap de 1000 réplicas, mostrando valores superiores al punto de corte (50%) de los haplotipos caracterizados para L. longipalpis y las secuencias registradas por Azpurua et al. (2010).
Discusión
Los resultados indican que el fragmento de Citocromo oxidasa I usado como código de barras para la identificación taxonómica de especies, permite discriminar haplotipos de L. longipalpis de secuencias pertenecientes a L. gomezi, L.cruciata y L. bifoliata ubicadas dentro del subgénero Lutzomyia, presentando un patrón de variación nucleotídica a lo largo de la secuencia, que posibilita la rápida identificación molecular de especímenes colectados en focos de transmisión de leishmaniasis.
Estudios de "código de barras" han sido aplicados a otros insectos de interés médico y veterinario determinando un patrón de variación intra-especifico no mayor al 4%: Culicidae (0-1,8%) (Kumar et al. 2007), Tabanidae (0-3,3%) (Cywinska et al. 2010) y Simuliidae (0-3,84%) (Rivera y Currie 2009). En Phlebotominae el análisis de las secuencias registradas por Azpurua et al. (2010) revela distancias intraespecie no mayores a 4% para L. trinidadensis, L. gomezi y L. panamensis, en nuestro estudio encontramos porcentajes de divergencia mayores (5-7%) para individuos de L. longipalpis provenientes de varias localidades de Colombia, que presentan diferentes condiciones locales a nivel ecológico y geográfico (González et al. 2006), así como una baja capacidad de dispersión del insecto a nivel local (< 1 km) (Morrison et al. 1993), evidenciando procesos adaptativos que sugieren la probable existencia de un complejo de especies en Colombia, como se ha demostrado en Brasil (Bauzer et al. 2007) y Venezuela (Arrivillaga et al. 2000).
Para L. longipalpis se registraron haplotipos únicos por localidad, reflejando una alta segregación geográfica, lo cual fue confirmado por los agrupamientos en el dendrograma de Neighbor-Joining, con haplogrupos relacionados (Neiva y El Callejón) y divergentes (Girón / Neiva + El Callejón). Esta relación evolutiva cercana entre individuos de Neiva y El Callejón fue también registrada por Uribe et al. (2001) al evaluar las relaciones filogenéticas de especimenes del intervalo de distribución de L. longipalpis mediante el gen ND4. Las distancias genéticas estimadas y las relaciones evolutivas inferidas (Girón-Neiva + El Callejón) con los haplotipos de COI son similares a los haplotipos caracterizados con ND4 (Uribe et al. 2001) y COI (Arrivillaga et al. 2002), pero difieren de los resultados publicados por Lanzaro et al. (1998), al obtener bajas distancias de Nei a partir de isoenzimas con individuos de Neiva, Melgar (Cundinamarca) y Palo Alto (Santander), concluyendo que L. longipalpis en Colombia constituye una sola especie con baja variabilidad genética.
El nivel de divergencia inter-específico presentó un intervalo de valores (12-18%) no solapado con los obtenidos en L. longipalpis, probando la eficiencia del marcador para diferenciar especies; así mismo, es destacable la alta divergencia entre haplotipos de L. longipalpis de diferentes localidades, haciendo este marcador atractivo para futuros estudios en esta especie. En este sentido, sería importante construir una librería con secuencias de COI de L. longipalpis, incluyendo el mayor número de localidades donde se ha registrado la especie, y en particular, aquellas diferentes a bosque seco tropical (Norcasia, PECET, datos no publicados) entre otras (López et al. 1996; González et al. 2006), esto permitiría evaluar la utilidad de la secuencia "códigos de barra" para esta especie en el rango completo de su distribución y precisar el aspecto de la existencia del complejo de especies L. longipalpis para el país.
Footnotes
Agradecimientos
Los autores agradecen a la Universidad Nacional de Colombia-Sede Medellín por la financiación de este trabajo enmarcado en el Proyecto DIME QUIPU 20101009543 y al Dr Charles Porter del CDC por su apoyo constante y asesoría.