
Abstract
The positron emission tomography (PET) ligand [11C]carfentanil is a selective agonist for μ-opioid receptors and has been used for studying μ-opioid receptors in the human brain. However, it is unknown if [11C]carfentanil binding differentiates between subtype receptors μ1 and μ2. In this study, we investigated whether μ1 and μ2 can be studied separately through receptor subtype–selective inhibition of [11C]carfentanil by pharmacologic intervention. [11C]Carfentanil binding characteristics on rat brain sections were assessed either alone or in the presence of the μ-receptor inhibitor cyprodime or the μ1-specific inhibitor naloxonazine. [11C]Carfentanil binding in the living rat brain was similarly studied by small animal PET/computed tomography during baseline conditions or following displacement by cyprodime or naloxonazine. Autoradiography binding studies on rat brain sections demonstrated that [11C]carfentanil has higher affinity and binding potential for μ1 than for μ2. [11C]Carfentanil binding to μ2 in vivo could not be detected following specific blocking of μ1, as predicted from the low binding potential for μ2 as measured in vitro. [11C]Carfentanil binding is preferential for μ1 compared to μ2 in vitro and in vivo. Clinical studies employing [11C]carfentanil are therefore likely biased to measure μ1 rather than μ2.
THE OPIOID RECEPTORS have been an important target for analgesics since the nineteenth century following the widespread use of morphine. They are mediators of the powerful analgesic effects by both endogenous opiate peptides and natural alkaloids (e.g., morphine) and synthetic analogues. The μ-opioid receptors are of general interest as morphine and similar opioids with analgesic effects, such as oxycodone, have high affinity to this subtype. The μ-opioid receptors can be further divided into subtypes, μ1 and μ2, in addition to the recently discovered μ3. 1
The μ1-opioid receptor has been associated with the desired analgesic effect, whereas μ2 receptors mediate the unwanted side effects of morphine-based drugs, such as respiratory depression and reduced motility of the gastrointestinal tract.2,3 For this reason, it is imperative to understand the difference in pharmacology, structure, and biodistribution between these receptor subtypes.
Methodologies for studies of in vivo distribution and the pharmacokinetics/pharmacodynamics (PK/PD) of ligands interacting with the μ1- and μ2-opioid receptor subtypes are therefore of interest. The positron emission tomography (PET) ligand [11C]carfentanil is a selective agonist for μ-opioid receptors and has been used for studying all μ-opioid receptor subtypes in aggregate in the human brain, 4 for example, during disease progression 5 and in states associated with drug dependency. 6 However, not much is known about whether [11C]carfentanil binding differentiates between subtype receptors μ1 and μ2.
Since the receptor binding of [11C]carfentanil is reversible, it can be used to indirectly study the pharmacodynamics of μ- and potentially μ1- or μ2-selective agonists or antagonists in vivo by observing their displacement of [11C] carfentanil in vivo. 7 In addition, important PK/PD parameters, such as the rate of onset of binding (kon) and degree of receptor occupancy of unlabeled μ-, μ1-, or μ2-selective compounds, can be quantified in the human brain.
In this study, we investigated whether the regional distributions of μ1 and μ2 could be studied separately through selective displacement of [11C]carfentanil, both in vitro on rat brain sections and in the living rat brain by small animal PET/computed tomography (CT).
Method and Materials
Radiochemistry
[11C]Carfentanil was synthesized as described previously, 8 modified to the synthesis equipment at the PET Centre at Uppsala University Hospital.
Pharmaceuticals
Cyprodime (Tocris Biosciences, Bristol, UK) was used as a selective general μ-opioid receptor antagonist. 9 A 5 mM stock solution was prepared by dissolving cyprodime in 30% hydroxypropyl-β-cyclodextrin (Kleptose, 300 mg/mL, Apoteket, Stockholm, Sweden).
Naloxonazine (Tocris Biosciences, Bristol, UK), a derivate of naloxone, is a selective antagonist to μ1. 10 A 1 mM stock solution was prepared by dissolving naloxonazine in phosphate-buffered saline (NaCl 137 mM, KCl 2.7 mM, Na2HPO4 10 mM, KH2PO4 1.8 mM).
In Vitro Frozen Tissue Autoradiography
Freshly harvested rat brain (Sprague Dawley, male) was frozen to −80°C and processed into 20 μm sections by a microtome. To study tracer binding properties to the tissue, the sections were incubated in progressively higher concentrations (0-50 nM) of [11C]carfentanil (n = 5 batches in total) in 50 mM TRIS + 0.5 mm ethylenediaminetetraacetic acid (EDTA) for 40 minutes at room temperature. Nonspecific binding was assessed by adding 40 μm of the general μ-receptor antagonist cyprodime to the incubation buffer 10 minutes before tracer addition. Specific μ2-receptor binding was assessed by instead adding 0.5 μm of naloxonazine, which selectively inhibits the μ1 receptor. The dose was kept relatively low (but still 500 times above the expected subnanomolar Kd) in order not to inadvertently inhibit other μ-opioid receptors.
Tissue sections were then washed two times for 2.5 minutes in 50 mM TRIS + 0.5 mM EDTA at room temperature to remove excess tracer and then dried at 37°C for 5 minutes. The sections were then exposed against a phosphor-imager screen (Amersham Biosciences, Uppsala, Sweden) for 2 hours, digitalized using a PhosphorImager SI (Molecular Dynamics, Sunnyvale, CA), and analyzed using ImageQuant (Molecular Dynamics).
The data from all five experiments (two to three tracer concentrations in each experiment) were pooled, yielding 12 data points in total.
It was assumed that incubation with [11C]carfentanil only resulted in total binding to each section, that is, both receptor-specific binding of the μ1 and μ2 receptors and nonspecific binding. Coincubation with cyprodime (which is a general μ-receptor inhibitor) was assumed to block all μ receptor-mediated [11C]carfentanil binding, and the remaining binding thus represented nonspecific binding in each tissue. Finally, naloxonazine selectively inhibits μ1; thus, coincubation with [11C]carfentanil will yield autoradiography images, which represent μ2 binding in aggregate with nonspecific binding.
For each tracer concentration, it is now possible to calculate the following:
Binding to μ1 + μ2 = only [11C]carfentanil added (total binding) — [11C]carfentanil with cyprodime added (nonspecific binding) (Equation 1)
Binding to μ2 = [11C]carfentanil with naloxonazine added — [11C]carfentanil with cyprodime added (nonspecific binding) (Equation 2)
Binding to μ1 = (1) — (2) (Equation 3)
Calculation of binding to μ1, μ2, and both μ receptors in aggregate was performed for each data point as described above and plotted against the added concentration of [11C] carfentanil.
The tracer affinity (expressed as the dissociation constant Kd) for each receptor type and receptor density (Bmax) was determined by nonlinear regression of receptor-specific binding using GraphPad Prism 5 (San Diego, CA). The Kd was constrained to be shared between thalamus and cortex. The in vitro binding potential (BP) was calculated as the ratio Bmax/Kd.
In Vivo PET
Sprague Dawley rats (male, n = 11, weight 338 ± 24 g) were housed under standard laboratory conditions with free access to food and water. All handling and experiments were carried out in accordance with national guidelines and were approved by the local ethics committee for animal research.
Anesthesia was induced by 3.0% isoflurane in 450 mL/min of 50% medical air/50% O2. The animal was placed on the heated bed of a Triumph Trimodality PET/single-photon emission computed tomography (SPECT)/CT system (Gamma Medica Inc, Northridge, CA) to prevent hypothermia, where continuous anesthesia was maintained by 1.5 to 2.0% isoflurane through a face mask. The breathing rate and body temperature were monitored by an integrated physiologic monitoring system. The animal was positioned with the intra-aural line in the center of the transaxial field of view.
[11C]Carfentanil (n = 11, 6.2 ± 1.8 MBq corresponding to 0.08 ± 0.02 μg, or 0.24 ± 0.07 μg/kg of mass of carfentanil) in a maximum volume of 500 μL was administered as a singlebolus injection via a tail vein catheter, and the animal underwent a dynamic PET examination of the brain for 60 minutes in list mode, followed by a CT examination for 3 minutes (field of view = 8.0 cm).
The PET examinations were performed as baseline scans (n = 2), as displacement studies, or at different time points following intravenous pretreatment with one of two μ-receptor antagonists (see Table 1 for details). Cyprodime (selective general μ inhibitor, 3 mg/kg, n = 2) or naloxonazine (selective μ1 inhibitor, 1 mg/kg, n = 2) was thus administered 30 minutes following tracer injection to investigate the displacement of [11C]carfentanil. Additionally, naloxonazine was administered 1.5 hours (n = 1), 6 hours (n = 2), or 24 hours (n = 2) prior to [11C]carfentanil to investigate the time dependence on the blocking of μ1. It has previously been shown that doses below 10 to 20 mg/kg naloxonazine intravenously in rats produce μ1-selective binding in vivo. For the 1.5- to 24-hour time points, naloxonazine was administered under brief anesthesia (< 5 minutes). The animals were then allowed to wake up in each housing cage until being anesthetized again for the PET examination. Neither cyprodime (3 mg/kg) nor naloxonazine (1 mg/kg) induced any effects on animal physiology regarding temperature, breathing rate, or heart rate.
After the PET/CT examination, the animals were euthanized by an intravenous injection of 40 mg pentobarbital.
The PET data sets were reconstructed into 12 time frames (12 × 5 minutes) using a maximum likelihood expectation maximization (MLEM) three-dimensional algorithm (10 iterations) with corrections for scatter and random coincidences. The CT raw files were reconstructed using filtered backprojection and converted to DICOM. PET/CT data were analyzed using PMOD v3.13 (PMOD Technologies Ltd, Zurich, Switzerland). Volumes of interest were drawn manually on fused PET/CT images with support of a brain atlas to include thalamus, cortex, and cerebellum. [11C]Carfentanil kinetics in thalamus, cortex, and cerebellum was expressed as standardized uptake values (SUVs), corrected for the amount of administered tracer (in MBq) and animal weight. Then uptake in the target tissues thalamus and cortex was expressed as a tissue to reference tissue ratio, using the cerebellum as the reference region being devoid of μ receptor—mediated binding. 11 The results on group levels are reported as means ± SEM.
Experimental Design of the In Vivo Imaging Study
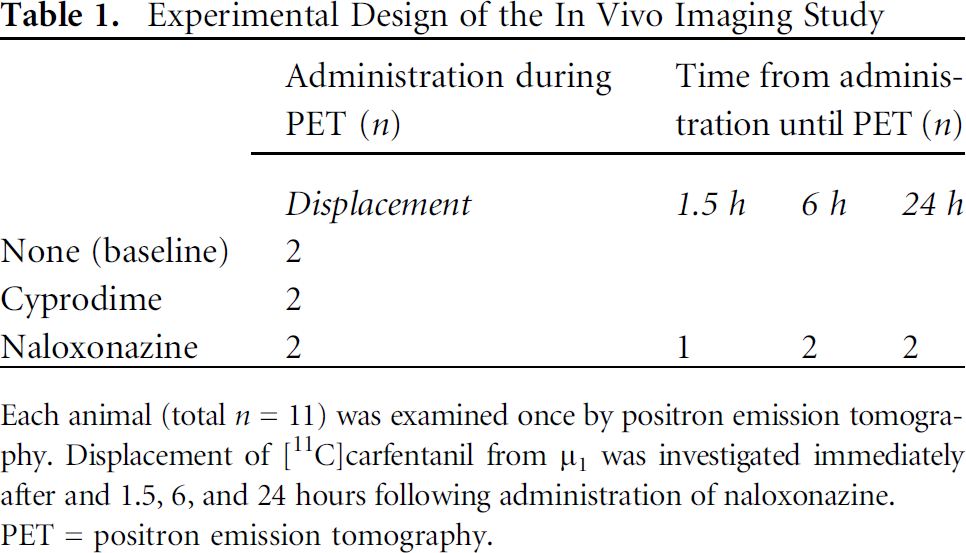
Each animal (total n = 11) was examined once by positron emission tomography. Displacement of [11C]carfentanil from µ1 was investigated immediately after and 1.5, 6, and 24 hours following administration of naloxonazine.
PET = positron emission tomography.
Results
Radiochemistry
The radiochemical purity of [11C]carfentanil (n = 9) was > 96%, and the specific radioactivity was between 7 and 111 GBq/μmol (on average, 45.8 GBq/mmol) at the end of synthesis.
In Vitro Frozen Tissue Autoradiography
High [11C]carfentanil binding was found in brain regions with known high expression of μ-opioid receptors such as thalamus, amygdala, cortex, and habenula (Figure 1). Coincubation with 40 μm cyprodime abolished all [11C]carfentanil binding, showing that tracer binding was μ receptor specific. Coincubation with 0.5 μm naloxonazine blocks only the μ1-receptor subtype; subsequently, the [11C]carfentanil binding in brain was almost, but not entirely, abolished. The remaining [11C]carfentanil most likely consists of μ2-mediated binding.

Distribution of [11C]carfentanil (2.8 nM) binding in rat brain sections (A), where 1 = cerebral cortex, 2 = thalamus, 3 = amygdala, and 4 = habenula. Coincubation with the μ1 receptor—specific antagonist naloxonazine inhibits the majority of, but not all, [11C]carfentanil binding (B). Only binding to μ2 receptor remains. Coincubation with the general μ-opioid receptor antagonist cyprodime abolishes nearly all [11C]carfentanil binding (C). The figure illustrates the regional binding at this specific concentration, 2.8 nM, of [11C]carfentanil.
[11C]Carfentanil exhibited saturable binding to μ receptors (as assessed by cyprodime) in thalamus with a Kd power of 10 above nanomolar (Figure 2, A—C, and Table 2). Binding in cortex was also saturable but with slightly lower Bmax (see Figure 2, D and E, and Table 2).
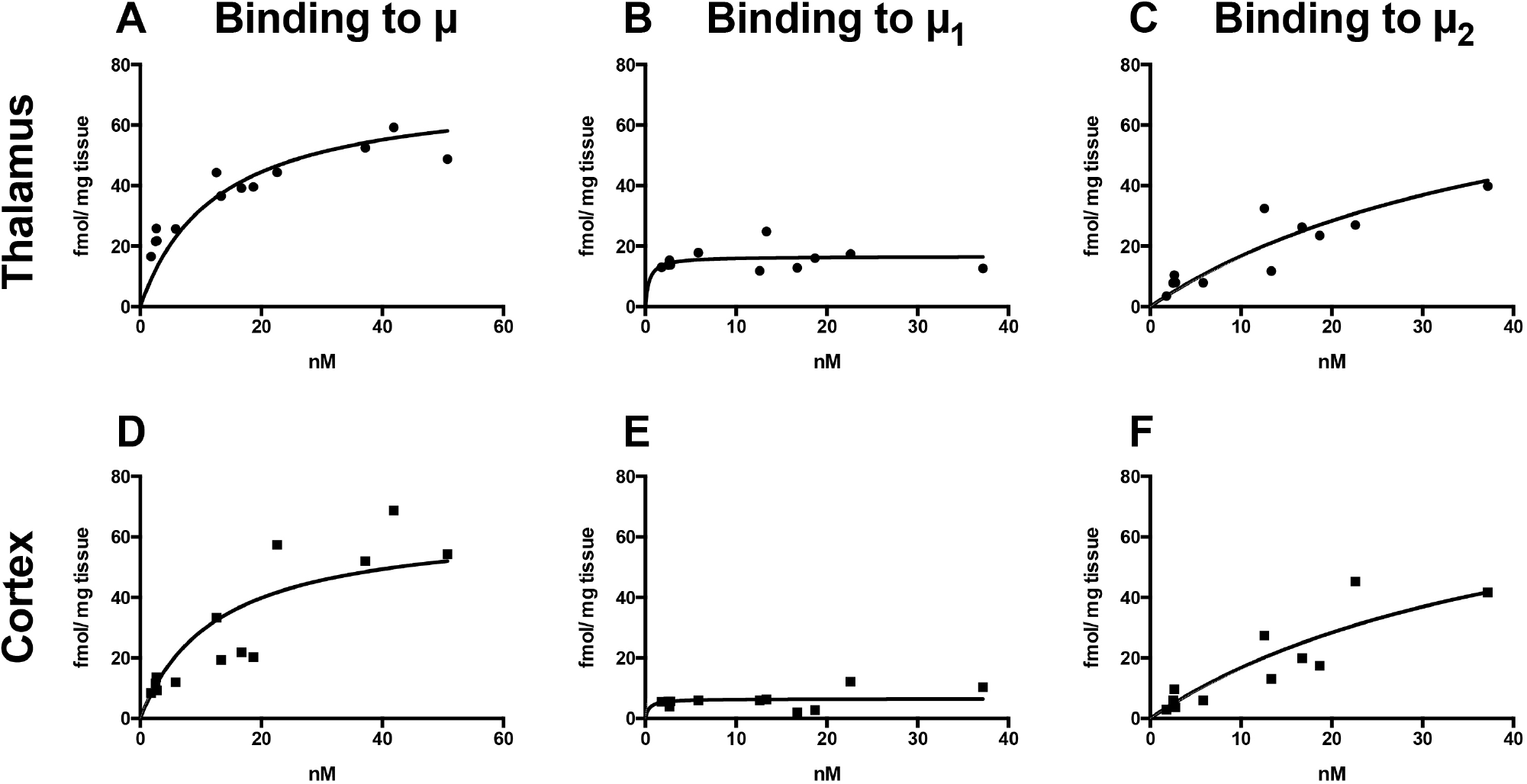
Autoradiographic saturation binding studies in thalamus (A—C) and cortex (D—F) in rat brain sections over [11C]carfentanil concentrations ranging from 0 to 50 nM. Binding to all μ receptors was assessed by coincubation with cyprodime (A, D). Binding to μ2 receptors was assessed by coincubation with naloxonazine (C, F). Binding to μ1 receptors was then calculated by subtracting μ2 binding from μ binding (B, E).
The μ1-specific binding of [11C]carfentanil was calculated by subtracting μ2-specific binding from the binding to all μ receptors as described in detail above. [11C]Carfentanil had lower affinity for μ2 compared to μ1, displaying only partial saturation for receptor concentrations up to 40 nM (see Table 2). BP for μ2 was similar in thalamus and cortex. The BP in thalamus was 20.1 and 7.9 times higher for μ1 compared to μ2 in thalamus and cortex, respectively.
In Vivo PET
Tracer uptake (expressed as SUV) was highest in opioid receptor—rich thalamus and low in cerebellum following administration of [11C]carfentanil only (Figure 3, A and E). There was a strong displacement of [11C]carfentanil in thalamus and, to a lesser extent, cortex, but not cerebellum, when cyprodime or naloxonazine was administered intravenously 30 minutes after tracer (Figure 3, B, C, E, and F). This indicates that cerebellum is devoid of receptor-mediated [11C]carfentanil binding and was therefore used as a reference region for further analysis.

Representative time-activity curves (expressed as SUV) in rat brain following injection of [11C]carfentanil as baseline (A) or displacement by cyprodime (B) or naloxonazine (C). Black arrows show the time point for the beginning of displacement. Panels (D) to (F) show transaxial projections from each PET examination, at the approximate level of the autoradiographic sections in Figure 1. White and red arrows indicate cortex and thalamus, respectively. The images are averages of 35 to 60 minutes following tracer injection, equivalent to 5 to 30 minutes following injection of each inhibitor. The images are normalized to SUV = 1 to allow for comparison between baseline conditions (D) and displacement by cyprodime (E) or naloxonazine (F). SUV = standardized uptake values.
[11C]Carfentanil accumulation in thalamus relative to cerebellum reached a plateau after 20 minutes, followed by slow washout especially after 40 minutes (Figure 4A). Administration of the μ-blocking antagonist cyprodime 30 minutes into the PET examination showed a rapid decrease in retention in thalamus, corresponding to displacement of [11C]carfentanil from available μ receptors (Figure 4B).

PET imaging of μ-opioid receptors in thalamus in the living brain (A). Retention of [11C]carfentanil in thalamus was displaced by cyprodime (B) and, to a lesser degree, naloxonazine (C). Black arrows indicate the time point for the beginning of displacement. Naloxonazine-mediated blocking of [11C]carfentanil in thalamus persisted at 1.5 (D) and 6 (E) hours but was normalized after 24 hours (F). Baseline kinetics is shown in all panels as a black dotted line. Data are given as averages ± SEM.
Naloxonazine, on the other hand, exhibited a slower onset of displacement of [11C]carfentanil from μ1 receptors. (Figure 4C) in thalamus. However, [11C]carfentanil administration 1.5 hours after naloxonazine treatment displayed no appreciable binding in thalamus at all, > 95% reduction compared to baseline (Figure 4D). Six hours postadministration of naloxonazine, [11C]carfentanil binding was reduced with approximately 50% compared to baseline, and binding was completely normalized after 24 hours (Figure 4, E and F).
Bmax and Kd of [11C]Carfentanil to µ, µ1, and µ2 Receptors

In vitro BP (calculated as the ratio Bmax/Kd), was higher for µ1 than for µ2, indicating preferential binding to µ1 at tracer levels. In vitro BP was higher in thalamus than in cortex, consistent with the known higher expression of µ-opioid receptors in thalamus.
Bmax = total receptor density; BP = binding potential; Kd = dissociation constant.
The uptake [11C]carfentanil in cortex relative to cerebellum followed a similar pattern, but with lower magnitude of binding, approximately 50% compared to binding in thalamus (Figure 5A). Cyprodime exhibited rapid displacement of [11C]carfentanil in contrast to naloxonazine (Figure 5, B and C). However, the binding of naloxonazine 1.5 hours postadministration completely inhibited [11C] carfentanil binding (Figure 5D). Similar to in thalamus, binding was increased at 6 hours and normalized 24 hours following naloxonazine administration (Figure 5, E and F).

PET imaging of μ-opioid receptors in cerebral cortex in the living brain (A). Retention of [11C]carfentanil in cortex was somewhat displaced by cyprodime (B) and, to a minor degree, naloxonazine (C). Black arrows indicate the time point for the beginning of displacement. The bottom panels (D to F) show the naloxonazine-mediated effect on blocking of [11C]carfentanil in cortex for up to 24 hours. Baseline kinetics is shown in all panels as a black dotted line. Data are given as averages ± SEM.
Discussion
[11C]Carfentanil has provided valuable insight into the understanding of human physiology at the μ-receptor level relating to addiction to opiate drugs, 6 epilepsy, 5 and other phenomena, such as pain. 12 The complexity of the human opioid receptor systems has been progressively unraveled during the last decades, and what were collectively termed opioid receptors were subdivided into three opioid receptor subtypes: δ, κ, and μ. These were found to be closely related but to have distinctly different effects when activated by agonists or antagonized. Furthermore, as molecular biology has evolved, these subcategories have been further refined to include subtypes of each of the δ, κ, and μ receptors.
[11C]Carfentanil was originally developed as a receptor-specific ligand for in vivo studies of the μ-opioid receptor system in the human brain. 4 However, with increased understanding that the μ-receptor subtypes 1 and 2 have specific distinctive roles in the regulation of analgesia and sedation, it becomes apparent that it is desirable to develop methodologies to image the receptor subtypes individually. No μ1- or μ2-selective radiolabeled ligands have yet been reported. Furthermore, the preferential binding of [11C]carfentanil to either μ-receptor subtype has not previously been examined. Therefore, we aimed to test if [11C]carfentanil could be employed in this fashion in vivo by selectively inhibiting the m1 receptor using naloxonazine.
Our in vitro autoradiography saturation binding results demonstrate that [11C]carfentanil has a distinctly heterogeneous binding profile from μ1- and μ2-receptor subtypes. Binding to μ1 occurs with higher (> 100-fold) relative affinity than for μ2. The apparent affinity toμ receptors, which is just above the nanomolar range, thus consists of a mixture of very high affinity for μ1 and medium high affinity for μ2. Similarly, BP for μ1 was more than 20 times higher than for μ2 in thalamus.
Additionally, we attempted to selectively image the μ1 and μ2 receptors in the living rat brain by employing a technique similar to the in vitro autoradiography study. Selective blocking of the μ1 receptors by naloxonazine should in theory leave only μ2 receptors for [11C]carfentanil to interact with. However, we could only detect a slower displacement by using naloxonazine compared to cyprodime, and following 1.5 hours, no appreciable [11C]carfentanil binding was detected. It is conceivable that the slower displacement could be attributed to the remaining binding of [11C]carfentanil to m2 receptors, but given that imaging at later time points revealed no μ2 binding above background, it is more likely that this effect is due to a more gradual onset of binding for naloxonazine compared to cyprodime. The difference in onset of binding could also be a consequence of the higher dose of cyprodime given (3 mg/kg) in comparison to that of naloxonazine (1 mg/kg).
The low or immeasurable in vivo binding of [11C]carfentanil in the presence of naloxonazine likely reflects poorer affinity of [11C]carfentanil to μ2, based on the results from the in vitro autoradiography study. However, it could also be due to a lower abundance of μ2 receptors.
Given the large differences in in vitro BP (> 20-fold higher in thalamus), we interpret the in vivo results to mean that quantitative imaging of μ2 is not feasible even when selectively inhibiting available m1 receptors.
We observe that weak but detectable binding of [11C] carfentanil can be measured by autoradiography in thalamus also after blocking with naloxonazine (see Figure 1B), which seems to conflict with the PET imaging results (see Figure 4D). This is likely due to the lower sensitivity (orlargererror bars)of the imaging technique compared to the autoradiography technique.
The findings reported here are of interest if the results are translatable to the human situation because this means that previous and future clinical studies employing [11C]carfentanil will be biased to measure μ1 rather than μ2.
Footnotes
Acknowledgment
Ram Kumar Selvaraju, Veronika Asplund, and Beatrice Borg are acknowledged for excellent technical assistance.
Financial disclosure of authors and reviewers: None reported.