
Abstract
Homing is an inherent, complex, multistep process performed by cells such as human bone marrow mesenchymal stem cells (hMSCs) to travel from a distant location to inflamed or damaged tissue and tumors. This ability of hMSCs has been exploited as a tumortargeting strategy in cell-based cancer therapy. The purpose of this study was to investigate the applicability of 111In-oxine for tracking hMSCs in vivo by combining single-photon emission computed tomography (SPECT) and magnetic resonance imaging (MRI). 111In-labeled hMSCs (106 cells) were infused intraperitoneally in neuroblastoma-bearing mice, whereas a control group received a dose of free 111In-oxine. SPECT and MRI studies were performed 24 and 48 hours afterwards. Initially, the images showed similar activity in the abdomen in both controls and hMSC-injected animals. In general, abdominal activity decreases at 48 hours. hMSC-injected animals showed increased uptake in the tumor area at 48 hours, whereas the control group showed a low level of activity at 24 hours, which decreased at 48 hours. In conclusion, tracking 111In-labeled hMSCs combining SPECT and MRI is feasible and may be transferable to clinical research. The multimodal combination is essential to ensure appropriate interpretation of the images.
HUMAN MESENCHYMAL STEM CELLS (hMSCs) are present in bone marrow niches. They can differentiate into cells of the mesodermal lineage (e.g., osteoblasts, adipocytes, and chondrocytes) and have the intrinsic ability to respond by migrating to damaged or inflamed tissues.1,2 In this regard, hMSCs have been used to regenerate injured tissue (e.g., infarcted myocardium) in animals and patients.3,4 hMSCs are also capable of homing to several types of tumors, including glioma, carcinoma (breast, colon, and lung), leukemia, and many types of metastatic tumors. Depending on the tumor microenvironment, hMSCs can display antitumoral properties, such as decreasing cancer cell growth, or protumoral properties, such as promoting metastasis or suppression of the immune response.5,6
In this work, we assessed the ability of injected cells to actively home to tumors, which supports the use of hMSCs as therapeutic vehicles to deliver anticancer agents. Garcia-Castro and colleagues showed that hMSCs irradiated and infected with oncolytic adenoviruses (CELYVIR) can be used as an effective therapy against metastatic and refractory neuroblastoma in children. 7 A complete clinical response was documented in one case. In that study, the number of metastases decreased, suggesting adenovirus activity because of hMSC homing. In our study, we used an imaging-based animal model to verify that hMSCs migrate to the neuroblastoma. Our approach can thus be transferred to human studies.
Migration of hMSCs can be tracked using molecular imaging modalities. Besides genetic modification, hMSCs can be labeled with different radiotracers (99mTc-hexamethylpropyleneamine oxime [HMPAO], 111In-oxine, and [18F]fluorodeoxyglucose [FDG]), (ultrasmall) superparamagnetic iron oxide (SPIO) particles, and quantum dots. The use of the corresponding imaging modality provides noninvasive methods for tracking and quantifying the fate of hMSCs administered in vivo, although not all available procedures are applicable to humans.6,8 de Vries and colleagues demonstrated that magnetic resonance imaging (MRI) is at least as sensitive as scintigraphy for detecting dendritic cell migration after intranodal coinjection of an equal number of 111In-oxine- and SPIO-labeled cells in patients. 9 However, given that the use of SPIO compounds in humans is restricted because of safety concerns, transferring this approach from animal models will remain problematic until better methods of introducing SPIOs into nonphagocytic cells are developed.9,10
Although many strategies are able to label and track stem cells in vivo, 11 only 111In-oxine and 99mTc-HMPAO are currently approved as clinical procedures (indicated for the detection of infections and inflammation by labeling leukocytes). 12 One obvious disadvantage of nuclear imaging is its potential effect on cell viability and proliferation. On the other hand, nuclear imaging offers better sensitivity, tissue penetration, and quantitative properties than optical imaging and MRI. 111In-labeled cells have been used for many years to track leukocytes and thus localize inflammatory processes in patients. Chin and colleagues studied the biodistribution of 111In-oxine-labeled swine MSCs 14 days after administration. 13 111In-labeled MSCs have also been applied in several preclinical settings: myocardial accumulation after intravenous administration in a swine myocardial infarction model, 13 dynamic distribution to different organs after infusion through different portals in healthy rats, 14 and analyses of the effects of 111In-labeled MSCs on cell proliferation, differentiation, and viability.2,15 In contrast to 111In-oxine, 99mTc-HMPAO could be administered at higher doses to improve image quality. However, its radiolabeling efficiency is poor because its retention within the cells is lower than that of 111In-oxine. 16 The short half-life of FDG (2 hours) reduces its applications in longitudinal studies. 17 Some novel techniques, such as direct 89Zr-oxine labeling, have shown good preliminary results.18,19 Besides the improvement in spatial resolution from single-photon emission computed tomography (SPECT) to positron emission tomography (PET), 89Zr-oxine imaging also needs 10 times lower activity compared to 111In-oxine. However, the 89Zr-oxine labeling protocol has some disadvantages compared to 111In-oxine. 19 For instance, Meszaros and colleagues showed that 89Zr-oxine labeling efficiency was 2.5 times lower in 5T33 myeloma cells compared to 111In-oxine. 18
Over the last decade, MRI has been emerged as a powerful tool for preclinical cell tracking due to its high spatial resolution. In some scenarios, the anatomic resolution may render this technique more suitable than SPECT, when the cells are directly injected into the target area. 9 Nevertheless, with intravenous administration, MRI is not yet as sensitive as SPECT. 20
Multimodality imaging using separate scanners requires very careful transfer of the animal followed by image coregistration. Some authors used SPECT–computed tomography (CT) systems,15,20 although MRI provides much better anatomic detail. Alignment accuracy depends on the resolution of both modalities and on possible image artifacts. Our small-animal SPECT studies have a spatial resolution of about 1 mm3. To facilitate SPECT/MRI coregistration, we used an in-house multimodality bed combined with a semiautomated rigid registration method based on matching lines. 21 The approach resulted in target registration errors close to the SPECT spatial resolution (< 0.9 mm). In terms of accuracy, our registration results are consistent with those reported in the literature. 22 Despite the availability of several methods for coregistering SPECT and MRI images,22,23 we did not find references to coregistering SPECT and MRI in cell-tracking experiments, where activity at the target can be very low and accurate spatial registration is required. A hybrid system (SPECT- MRI or PET-MRI) would facilitate this kind of approach.
The purpose of our work was to use a multimodality approach combining the sensitivity of SPECT with the anatomic reliability of MRI to detect hMSC homing in neuroblastoma-bearing mice.
Material and Methods
Ethics Statement
All animal procedures were approved by the Animal Experimentation Ethics Committee of Hospital General Universitario Gregorio Marañón (ES280790000087) and were performed according to European Union directive (2010/63/EU) and national regulations (RD 53/2013). All human procedures were approved by the Clinical Research Ethics Committee of Hospital Universitario Nino Jesus (Madrid, Spain), and written informed consent was obtained from all participants (R-0030/09).
Cell Isolation, Culture, and Characterization
hMSCs were obtained following a previously described protocol 7 from three different donors. Briefly, mononuclear cells were isolated from bone marrow aspirates using Ficoll density centrifugation, resuspended in MSC medium (Dulbecco's Modified Eagle's Medium [DMEM] plus 10% fetal bovine serum), seeded into culture flasks at 3 × 104 cells/cm2, and allowed to adhere. After the fourth passage, a homogeneous hMSC culture was obtained and characterized according to the criteria of the International Society for Cellular Therapy. 24 Cells were frozen at 1.8 × 106 per milliliter after the fifth passage.
The human neuroblastoma cell line NB-1691 was derived from a neuroblastoma in a pediatric patient at Hospital Universitario Niño Jesús. Cells were cultured in monolayer at 37°C with 5% CO2 in DMEM (Lonza, Switzerland) supplemented with 10% fetal bovine serum (PAN-Biotech GmbH, Germany).
111In-Oxine Labeling
Cells were labeled following the protocol of Gildehaus and colleagues. 15 Briefly, cells were thawed and centrifuged at 1,800 rpm for 5 minutes, and the cell pellet was resuspended with 10 Bq/cell 111In-oxine in 1 mL of phosphate-buffered saline (PBS) and incubated for 20 minutes at 37°C. Cells were then washed twice with PBS, and the activity of the supernatants and cell pellet (after PBS washes) was measured in an activimeter (Capintec CRC, USA). The labeling yield was calculated by dividing cell activity by total activity (activity in cells and in supernatant). For administration, around 1 million 111In-labeled hMSCs per animal were separated and resuspended in 0.5 mL PBS with heparin (1 unit per 0.1 mL). The solution was visually inspected to avoid clumps or aggregates.
Animals
Orthotopic human tumor xenografts are an excellent model for predicting physiologic and treatment response in human tumors. 25 For this reason, we developed a xenograft model in NOD/SCID male immunodeficient mice. Nine animals received one subcutaneous injection of 2.5 × 106 NB-1691 cells (resuspended in 100 μL of PBS/injection) in the left hind flank. The NB-1691 cell line was selected because tumor-secreted chemokines are more similar to those secreted by human neuroblastoma tumors. 26
Three weeks after tumor implantation, seven mice received an intraperitoneal dose of 1 million 111In-labeled hMSCs (hMSC-injected animals) in 0.5 mL PBS. Two control animals received an equivalent intraperitoneal dose of free 111In-oxine. Both 111In-labeled hMSCs and free 111In-oxine were injected when labeling was finished to avoid cell damage or radiopharmaceutical degradation. hMSCs were injected intraperitoneally in the right upper abdominal quadrant to maximize the distance to the tumor.
SPECT/MRI
Multimodality imaging was performed with a small-animal SPECT scanner (mSPECT, MILabs, the Netherlands) and a preclinical MRI system (7T Biospin, Bruker, Germany). SPECT and MRI images were acquired 24 and 48 hours after administration of 111In-labeled hMSCs or free 111In-oxine. To coregister the SPECT and MRI images, each animal was placed on an in-house multimodal bed surrounded by three noncoplanar capillaries filled with a mixture of 99mTc and CuSO4, which was visible in both modalities. The use of different SPECT radionuclides for the fiducials allowed us to separately reconstruct the capillaries and animal images using the respective radioisotope photopeaks (140 keV for 99mTc and 171 + 245 keV for 111In). This procedure allows for good visualization of the capillaries while preventing spillover from the fiducials into the tumor region.
The SPECT acquisition parameters were an isotropic voxel size of 0.4 mm and an acquisition time of 1.5 to 2 hours. SPECT images were reconstructed using two-dimensional ordered subset expectation maximization (OSEM-2D) with 16 subsets and 1 iteration. The MRI was obtained immediately after completion of SPECT imaging. A coronal MRI was acquired using a turbo rapid acquisition with refocused echo (turboRARE) T2 sequence 27 with a volume coil and the following parameters: echo time 15.9 ms, repetition time 3,425 ms, field of view 6 × 4.13 cm2, matrix size 465 × 256, rare factor of 8, and an acquisition time of 5 minutes.
We intentionally sacrificed MRI quality to achieve a fast MRI acquisition on animals that had already spent 1.5 to 2 hours under anesthesia while the SPECT image was acquired. To prevent any animal displacement between modalities, MRI was performed without any animal vital sign monitoring (respiration, electrocardiography, or temperature). This prevented us from acquiring a respiratory gated study, which would have improved quality by reducing breathing artifacts.
SPECT/MRI Coregistration
The spatial rigid transformation to align MRI to SPECT images 21 was obtained by segmenting the capillaries, adjusting each capillary to a line, and matching the corresponding fiducials of both modalities following a method proposed by Pascau and colleagues, 28 which was based on minimizing the distance between corresponding segments of lines.
Semiquantitative Image Analysis
On the MRIs, regions of interest (ROI) were selected over the entire volume of the tumor and abdomen (five circular sections of 3 mm diameter) at each time point (Figure S1, online version only). These ROI were applied to the SPECT images to obtain the mean activity values of the entire volume of the ROI. Activity values were corrected for the radioactive decay.
111In Distribution by Organ
Organ activity was measured directly in two control animals and five hMSC-injected animals after the last imaging session. The animals were sacrificed, and organs, including the liver, lungs, kidneys, long bones (tibia and femur), testicles, and spleen, were harvested. These organs were weighed, and the activity was measured at the 111In photopeak in a well counter (Wallac Wizard 1480 counter, PerkinElmer, Spain). Lung uptake was used as a reference for calculating the relative specific activity of each organ.
Real-Time Quantitative Polymerase Chain Reaction
Real-time quantitative polymerase chain reaction (RT- qPCR) was used to confirm homing of the inoculated hMSCs in the neuroblastoma. Tumors were harvested and frozen from one control and two hMSC-injected animals after the last imaging session. Total ribonucleic acid (RNA) was isolated using the TRIzol method, according to the manufacturer's protocol (Life Technologies, Spain), and 2 μg was converted into complementary DNA (cDNA) using M-MLV Reverse Transcriptase (Life Technologies) with oligo dT primers. RT-qPCR was performed using the LightCycler 480 SYBR Green I Master (Roche Applied Science, Spain) with the primers listed below and analyzed using the LightCycler System (Roche Applied Science). The sequences for the human vimentin and glyceraldehyde 3- phosphate dehydrogenase (GAPDH) primers are as follows: vimentin, forward, TGTCCAAATCGATGTGGATGTTTC, and TTGTACCATTCTTCTGCCTCCTG reverse; GAPDH, forward GCCAAGGTCATCCATGACAACT, and reverse AGGGCCATCCACAGTCTTCTG. Three replicates were performed per cDNA sample along with “no template” controls. The specificity of amplification was confirmed by melting curve analysis. GAPDH expression was selected as a housekeeping gene and used as an internal control for normalization of vimentin messenger RNA expression.
Statistical Analysis
Differences in tumor uptake between the experimental groups were assessed by linear mixed-model analysis of variance. Data for organ activity distribution were compared using Student t-test. Differences were considered statistically significant for p < .05.
Results
Cell Labeling Efficiency
Incubation of hMSCs with PBS containing 10 Bq/cell of 111In-oxine for 20 minutes resulted in a mean ± standard deviation labeling efficiency of 71.5 ± 10.6%. The activity of 1 million cells was 3.3 ± 1.4 MBq. Trypan blue staining showed a survival of 80% immediately after labeling. We had previously tested labeling using other radiotracers with a shorter half-life, namely, FDG and 99mTc-HMPAO.17,29 Although FDG and 99mTc-HMPAO showed higher cell viability than 111In-oxine (93%, 88% and 80%, respectively), hMSCs labeling efficiency with these two radiotracers was lower (18.5% for FDG and 44.4% for 99mTc-HMPAO) compared with the labeling efficiency achieved with 111In-oxine (72%).
Evidence of Homing of hMSCs to the Tumor
SPECT and MRI images were acquired 24 and 48 hours after intraperitoneal administration of 111In-labeled hMSCs or administration of free 111In-oxine (controls). In MRI, abdominal structures are visible and the tumor is easily identified. In SPECT, however, the lack of anatomic references makes it almost impossible to locate the tumor area and detect specific uptake. After multimodality fusion, activity from the tumor area can be separated from the background thanks to the ROI drawn on the MRI. Figure 1 shows coronal and axial views of the fusion of registered SPECT and MRI images of an hMSC-injected animal and a control animal. At both time points, the images showed similar activity in the abdomen in both controls and hMSC- injected animals. In general, abdominal activity decreases at 48 hours (Figure 2A). hMSC-injected animals showed increased uptake in the tumor area at 48 hours (p = .02), whereas the control group showed a low level of activity at 24 hours (possibly just reflecting spillover from the abdominal cavity), which decreased at 48 hours (Figure 2B). The uptake percentage in the tumor area at 48 hours was higher in hMSC-injected animals than in controls.
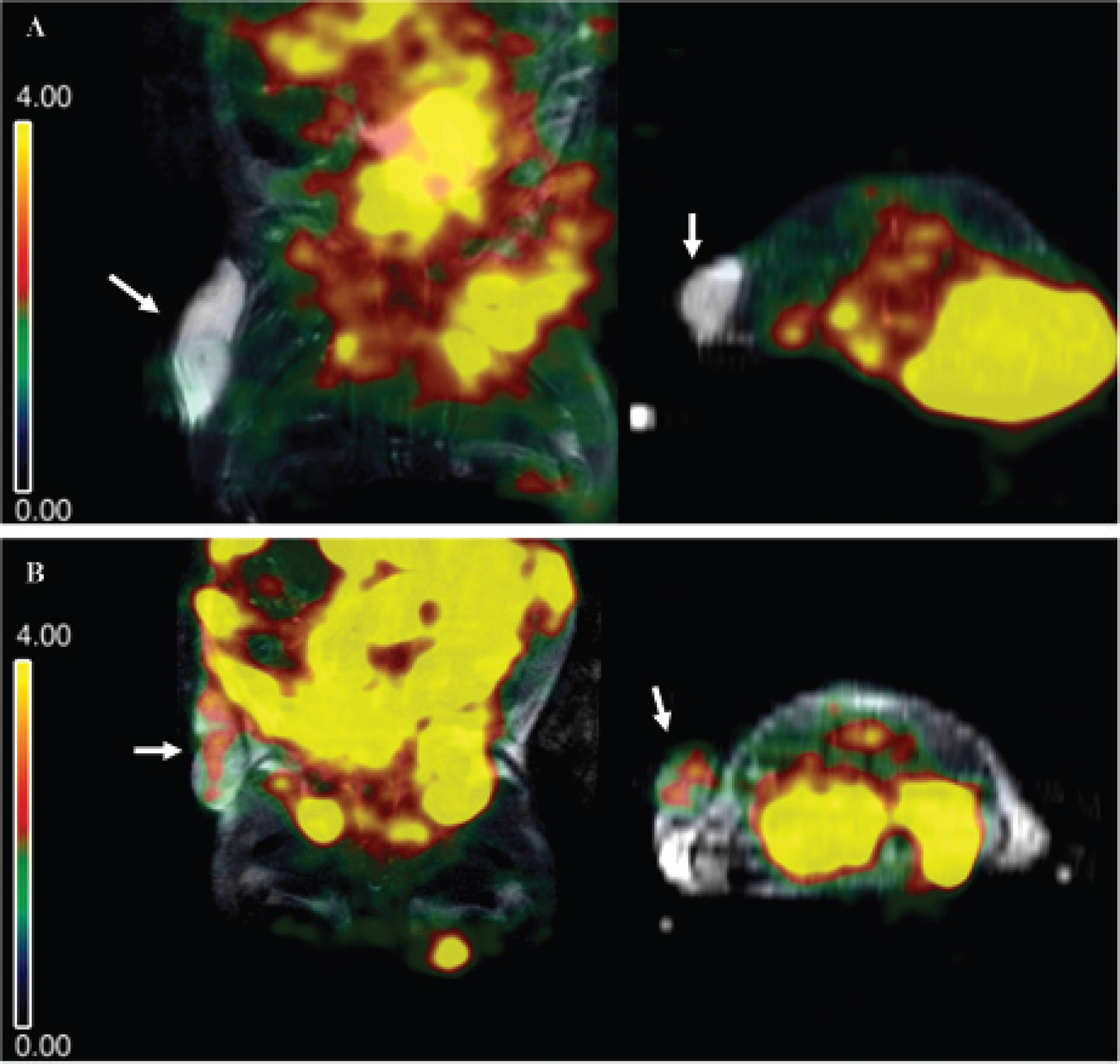
Coregistered MRI/SPECT images. Coronal and axial views of spatially coregistered MRI/SPECT images at 48 hours after intraperitoneal administration of 111In-oxine labeled human mesenchymal stem cells (hMSCs) or free 111In-oxine. Significant activity can be observed in the abdomen of both animals. A, The tumor in the control animal shows scant 111In activity. B, In the hMSC-injected animal, uptake can be observed inside the tumor area (white arrows).

Abdominal and tumor uptake measurements. Regions of interest (ROI) were selected in the MRI over the tumor and abdomen at each time point and applied to the SPECT images. Uptake at 48 hours is represented as a percentage of uptake at 24 hours (dotted line). A, In general, abdominal uptake decreases 48 hours after injection. B, Increased uptake was observed in the tumors of six of seven human mesenchymal stem cell (hMSC)-injected animals 48 hours after administration (dark bars), whereas activity in the control tumors decreased about 50%.
111In Organ Distribution
Organs were harvested immediately following the last scan, and activity was measured for 2 minutes; activity in each organ was corrected for organ weight and the decay factor. We used lung uptake as a reference value for calculating the relative specific activity of each organ. Forty-eight hours after injection, a number of cells seemed to be trapped in the liver and bone marrow, thus reducing the available number of homing cells (Figure S2, online version only). No significant difference in the distribution of activity was observed between controls and hMSC- injected animals.
Vimentin Expression
Our results revealed that the mesenchymal marker vimentin was expressed 4.5 ± 1.2 (mean ± SD) times more in the tumor tissue obtained from the hMSC-injected animals than in the tumor samples obtained from the control animals, indicating the presence of these cells. Control animals showed only mouse vimentin expression.
Discussion
In the present study, we developed a mouse model based on the neuroblastoma cell line NB-1691 to mimic the scenario used by Garcia-Castro and colleagues. 7 Although these authors reported a reduction in tumor metastasis in patients after infusion of adenovirus-infected hMSCs, they were not able to confirm hMSC tumor homing. We demonstrated that 111In-labeled human bone marrow–derived MSCs can home to neuroblastoma within 24 to 48 hours of intraperitoneal administration; to our knowledge, this work constitutes the first in vivo validation of such a hypothesis. We also showed that the multimodality approach, SPECT- MRI, is essential when localizing hMSCs.
The main criterion for choosing 111In-oxine as a tracer in this study was its long half-life, which allowed us to track cells for up to 48 hours. We did not assess proliferation and differentiation because in the cited clinical trial, hMSCs were used as a vehicle to deliver adenovirus, 7 whose replication cycle kills the hMSCs in 48 to 72 hours. Furthermore, several authors detected no hMSC damage at the dose we used (10 Bq per cell) within the first 48 hours after labeling.2,15 Gholamrezanezhad and colleagues found that the effect of 111In-oxine on hMSC viability is both time and dose dependent. 30 They reported a viability reduction of about 10% 48 hours after labeling with a dose of 7.16 MBq/106cells, almost two times the dose we used in our study (3.3 ± 1.4 MBq/106cells). In contrast, other authors reported cell damage 48 hours after labeling murine hematopoietic progenitor cells even at low doses (0.1 MBq/106cells).31,32 In summary, these studies show that cell damage induced by 111In labeling depends on both dose and cell type. Our results showed that labeling hMSCs with a dose of 10 Bq/cell of 111In-oxine does not destroy cell migration mechanisms within the first 48 hours. Although our hMSC viability immediately after labeling was reduced (80%), this may have been due to several causes besides 111In damage. It is well known that freezing and defrosting induce some cell damage. The mean cell viability just after defrosting has been previously reported to be about 85%.33,34 Furthermore, our labeling protocol led to an additional cell viability reduction of 3% due to washes and centrifugations (data not shown).
We incidentally observed that the use of thawed cells increased labeling efficiency from 52% to about 72%, probably because we avoided cell detachment from the flask before incubation with 111In-oxine. To confirm this finding, it would be necessary to further investigate differences in labeling efficiency between cultured and thawed cells using the same number of cell passages and the same hMSC donor.
Although the intraperitoneal route should ensure that all activity detected in the tumor was caused by migration of hMSCs, the activity from the testicles and abdomen was too close to the tumor area and produced an inconvenient spillover that hampered image interpretation. Kraitchman and colleagues found similar problems when they studied cell migration in animals with myocardial infarction. 20 For this reason, the availability of an MRI anatomic template proved to be critical for properly identifying the different regions. The time dynamics of ROI activity ensured that uptake was indeed due to hMSC tumor homing as 111In activity in the tumor 48 hours after administration increased in only six of seven hMSC-injected animals and decreased in controls. Although neither radiolabeling nor MRI labeling can discriminate live from dead cells, 35 our findings suggest that some hMSCs homed to the tumor within 24 to 48 hours after administration, meaning that hMSCs were alive in this time window. Although hMSC homing may be visualized a few hours after transplantation, 36 their final number in the target area depends on both time and the administration route. Barbash and colleagues reported that less than 1% of the cells migrated to the target area 4 hours after injection in the left ventricular cavity, whereas after intravenous administration, cell migration was four times lower at the same time. 37 Systemic administration is known to require a longer time window to enable visualization of cell homing. 31 For these reasons, we decided to acquire the images 24 and 48 hours after administration.
In our work, SPECT findings were corroborated by RT- qPCR results. Due to its high sensitivity, RT-qPCR has proven crucial in some clinical trials to detect transplanted MSC-specific signals.38,39 hMSC-injected animals showed higher vimentin expression than control animals. Although NB-1691 is a human line, after implantation, tumors are infiltrated with multiple mouse cell types, including fibroblasts and MSCs. Control tumors showed some degree of vimentin expression, probably owing to infiltration by mouse cells and the fact that human and mouse vimentin genes are highly homologous, thus making it difficult to design specific primers for each gene. Validation of vimentin primers used in this study was previously carried out in our laboratory as an internal control (data not shown). However, the higher level of vimentin expression in tumors obtained from hMSC-injected animals could be explained only by the fact that hMSCs migrated from the peritoneal cavity and homed to these tumors.
In the present study, hMSCs were injected intraperitoneally to avoid accumulation in the lungs because these cells are relatively large and express adhesion molecules.37,40 Our approach is supported by several previous works. Gao and colleagues reported that rat MSCs labeled with 111In-oxine accumulated primarily in the lungs and liver 15 minutes after intravenous or intra-arterial administration, whereas after intraperitoneal administration, activity appeared in the liver, testicles, spleen, and kidneys. 14 Similar results were reported by Rosado-de-Castro and colleagues 24 hours after intravenous or intra-arterial administration of 99mTc-labeled hMSCs in patients. 41 Cell trapping by the reticuloendothelial system seems to be the major cause of hMSC loss, regardless of the administration method, because it comprises phagocytic cells capable of engulfing and ingesting foreign cells and bacteria, as reported elsewhere. 42 We also observed high 111In activity in the testicular area in some animals 48 hours after administration and found this activity to be similar to that of the kidneys and higher than that of the liver (see Figure S2, online version only). We cannot be sure whether the cells trapped in these organs were dead or alive. It is possible that in some organs, such as the liver, both situations coexist. Brenner and colleagues reported an increased uptake in liver and kidney due to 111In released from the cells over time. 31 Although radiolabeling cannot discriminate live from dead cells, the 111In organ uptake found in our work is consistent with previous literature.13,37,41 These organs have been identified as the major site for homing of immunocompetent cells. 31 Nevertheless, bioluminescent MSC studies showed cell migration to the liver and spleen 24 hours after intravenous administration. Images after intraperitoneal administration indicated initial dissemination throughout the peritoneal cavity, followed by specific localization on day 7 in tumor-bearing animals and disappearance in control animals. On day 14, bioluminescent activity was localized only to sites of visible tumor. 43
Clinical neuroblastoma most frequently originates in one of the adrenal glands and also develops in nerve tissues in the neck, chest, abdomen, or pelvis. The most typical sites of neuroblastoma metastasis are bone marrow and bone. Specifically, the incidence of metastatic sites at neuroblastoma diagnosis is 70% in bone marrow, 55% in bone, 31% in lymph nodes, 29% in liver, 18% in intracranial and orbital sites, 3.5% in lung, and 0.5% in the central nervous system.44,45 The tumor of our xenograft model was far below the high nonspecific uptake showed in Figure 1, which may hamper hMSC migration in patients with abdomen or pelvis lesions. However, bone marrow and bone do not suffer as much from the problem of nonspecific signal. Thus, the combination of techniques proposed in this article may have a clinical application in most cases of neuroblastoma metastasis, although not in all patients, due to the areas with the nonspecific uptake problems cited above.
After intraperitoneal administration, it is possible that liquid flows inside the peritoneal cavity and accumulates in the scrotum. Combined with reticuloendothelial retention, this accumulation could further reduce the number of hMSCs able to home to the tumor. The results of cardiac animal studies show that only 1% of intravenously injected stem cells home to the heart; however, this amount can increase to 2% if cells are injected into the left ventricular cavity. 37 Hofmann and colleagues found similar results after intracoronary delivery of FDG-labeled bone marrow cells in patients with acute myocardial infarction, although no myocardial activity was observed after intravenous administration. 36 As mentioned above, we selected intraperitoneal administration of hMSCs to reduce or prevent lung cell trapping, as would occur with intravenous administration. Although intra-arterial administration also avoids lung trapping, it has been reported that it may lead to microvascular occlusions, 46 which may compromise the survival of the subject under study and might also attract the hMSCs, perhaps biasing the study. On the other hand, intraperitoneal administration produced high accumulation of cells in the peritoneal cavity and led to an inconvenient spillover into the tumor area. Perhaps injection of cells into the left ventricular cavity would be a better choice for future work because lung retention is also reduced and the number of cells able to migrate and colonize injured tissue is expected to be higher than with intravenous routes. 37 However, the main drawback of the left ventricular cavity administration is that it requires very specific techniques (such as echocardiography) to guide the procedure and involves significant survival risk in small animals.
In our study, each animal received around 3.3 ± 1.4 MBq of 111In-oxine per million cells, which is usually the lowest amount of cells injected for clinical therapeutic studies and almost the minimum 111In activity required in pediatric studies (patients less than 1 year old), thus making it possible to transfer this approach to clinical research.
Our study was limited by the small sample size, which does not enable a proper statistical analysis. This is the reason why in this work we do not emphasize the statistical analysis: the principal results are based on the image interpretation and semiquantitative results. No attempt was performed to calculate absolute activity values for two reasons: (1) peritoneal uptake differed greatly from animal to animal, and this prevented us from choosing any common reference region or organ, and (2) in contrast to PET, it is almost impossible to obtain reliable calibration factors in SPECT. 47 In addition, the use of cells from different donors introduces a certain degree of variability owing to possible differences in migration activity and labeling efficiency. Although our multimodal bed system worked properly for this mouse model, we cannot guarantee the absence of slight organ movements. Also, the quality of 111In imaging is affected by the fact that it has two photopeaks (171 and 245 keV) because higher energy radiation is more difficult to collimate. Finally, given the small number of hMSCs that homed to the tumor, RT- qPCR was chosen as a validation method because it is a quantitative method and more sensitive than histology.
In conclusion, we were able to detect homing of hMSCs to a neuroblastoma at 24 and 48 hours in a mouse model using coregistration of SPECT and MRI images, which proved to be essential. Our results were confirmed by RT- qPCR. The procedure can be used to monitor cell therapy provided that the activity of 111In-labeled hMSCs does not exceed the limits established for pediatric patients.
Footnotes
Acknowledgments
We thank Alexandra de Francisco and Yolanda Sierra for their excellent work with the animal preparation and imaging protocols and Dr. A. Pérez-Martínez (Hospital Universitario Niño Jesús, Madrid, Spain) for providing the neuroblastoma cell line.
Financial disclosure of authors: This work was funded in part by grants from Ministerio de Economía y Competitividad (PLE2009-0115), Red Tematica de Investigacion Cooperativa en Cancer (RTICC/ISCIII; RD12/0036/0027), the Madrid Regional Government (S-BIO-0204-2006–MesenCAM and P2010/BMD-2420-CellCAM), and the Ministerio de Ciencia e Innovación (CEN-20101014 and TEC-2010-21619-C04-01).
Financial disclosure of reviewers: None reported.