
Abstract
Excitotoxicity leads to an inflammatory reaction involving an overexpression of: translocator protein 18 kDa (TSPO) in cerebral microglia and astrocytes. Therefore, we performed ex vivo explorations with [125]-CLINDE, a TSPO-specific radioligand, to follow the time course of TSPO expression, in parallel with lesion progression, over 90 days after induction of cerebral excitotoxicity in rats intrastriatally injected with quinolinic acid. Biodistribution data showed a significant increase in CLINDE uptake on the injured side from 1 days postlesion (dpl); the maximal striatal binding values evidenced a plateau between 7 and 30 dpl. [125I]-CLINDE binding was displaced from the lesion by PK11195, suggesting TSPO specificity. These results were confirmed by ex vivo autoradiography. Combined immunohistochemical studies showed a marked increase in microglial expression in the lesion, peaking at 14 dpl, and astrocytic reactivity enhanced at 7 and 14 dpl, whereas a prominent neuronal cell loss was observed. At 90 dpl, CLINDE binding and immunoreactivity targeting activated microglia, astrogliosis, and neuronal cell density returned to a basal level. These results show that both neuroinflammation and neuronal loss profiles occurred concomitantly and appeared to be transitory processes. These findings provide the possibility of a therapeutic temporal window to compare the differential effects of antiinflammatory treatments in slowing down neurodegeneration in this rodent model, with potential applications to humans.
EXCITOTOXICITY OCCURS due to the overstimulation of N-methyl-D-aspartate (NMDA) receptors by glutamate and leads to increased calcium influx and production of free radicals, which can trigger a cascade of events, ultimately leading to neuronal death. 1 Therefore, excitotoxicity is recognized as an important biological process involved in neuronal death in a range of brain disorders and, in particular, neurodegenerative diseases.2,3 Quinolinic acid (QA) is an endogenous NMDA receptor agonist, and a metabolite of tryptophan, that is found in normal subjects as a byproduct of the kynurenine pathway, leading to the synthesis of the essential cofactors nicotinic acid and nicotinamide adenine dinucleotide. The unilateral intrastriatal injection of QA induces excitotoxicity via NMDA receptor activation and is considered an animal model mimicking early stages of Huntington disease as it reproduces some biochemical, behavioral, and pathologic features of the disease in rodents and nonhuman primates.4–6 Since the emergence of transgenic models of Huntington disease,7,8 the QA-induced striatal lesion became more relevant as a model of excitotoxicity. To date, this model is still widely used in studies assessing new therapeutic approaches9,10 and has been shown to produce large lesions accompanied by an inflammatory response involving increased microglial activation, which is directly associated with the degree of brain damage.3,11 Microglia are the resident immune cells of the brain that, under normal conditions, display a ramified morphology with small cell bodies and long, thin processes. The activation of microglia, following pathologic changes in the brain such as excitotoxic injury, includes changes in their morphology, adopting an amoeboid-like shape, migration toward the lesion site, proliferation, and acquisition of new functions in injured tissue, including the capacity to express and release a wide variety of proinflammatory molecules and to phagocytose dead cells. 12 The role of these activated microglial cells appears to be variable, with both harmful and beneficial effects on brain function (a currently frequently debated subject13,14), and it has been proposed that prolonged activation of microglia could contribute to the pathogenesis of neurodegenerative diseases.
One of the earliest molecules to be overexpressed during the microglial activation process is the translocator protein 18 kDa (TSPO), formerly known as the peripheral benzodiazepine receptor. 15 TSPO is a component of a multimeric protein complex closely associated with a voltage-dependent anion channel and an adenine nucleotide carrier. 16 TSPO is located on the outer membrane of the mitochondria and is widely distributed in peripheral organs. Although TSPO is minimally expressed in the healthy brain, its basal expression dramatically increases in several neurodegenerative and acute brain disorders, thus providing a sensitive biomarker for imaging microglial activation associated with neuroinflammation. 17
Molecular imaging has the capability of providing functional data with high resolution and sensitivity for following pathology and neurodegeneration noninvasively in the central nervous system. These can elucidate the temporal and spatial evolutions of the toxin-induced neuropathology, which are important determinants of animal model studies. 18 The current development of TSPO high-affinity radioligand allowing monitoring of the neuroinflammation process should be valuable for this purpose. Thus, the QA striatal lesion model has been widely investigated by molecular imaging.19–22 However, a comprehensive longitudinal molecular imaging study has not been performed yet on the QA model, although previous work has shown the evolution of the lesion until 60 days after surgery, but only referring to four time points, 23 although we have studied QA levels in a dose-response manner at one time point. 19 Indeed, a longitudinal study of links between neuroinflammation and neurodegeneration would improve understanding of the physiopathologic mechanism and accessibility to early diagnosis and/or new therapeutic approaches. Therefore, we aimed to characterize the longitudinal frame of this QA model as a prerequisite for the upcoming evaluation of new treatments modulating neuroinflammation. In that respect, we used the TSPO high-affinity ligand, 6-chloro-2- (4′iodophenyl)-3-(N,N-diethyl)-imidazo[1,2-a]pyridine-3- acetamide or CLINDE, to explore longitudinally expression of activated microglia in QA-lesioned rats. It has previously been shown that CLINDE was able to detect in vivo inflammatory processes characterized by increased density of TSPO in several preclinical models of neuroinflammation, including experimental autoimmune encephalomyelitis, 24 cerebral ischemia, 25 and temporal lobe epilepsy. 26 In the present work, we performed serial ex vivo cerebral biodistribution studies with [125I]-CLINDE to investigate the spatial and temporal density of TSPO at several time points (1, 4, 7, 14, 30, 60, and 90 days) after unilateral QA lesion in the rat striatum. This quantitative evaluation was complemented in parallel by ex vivo autoradiography, and the relevance of our findings was supported with immunohistochemistry data targeting neuronal survival, microglial activation, and astrogliosis.
Materials and Methods
Radiochemical Synthesis of [125I]-CLINDE
The radioligand [125I]-CLINDE was prepared as previously described, 19 with minor modifications. Briefly, the tributylin precursor (100 μg) in acetic acid (200 μL) was treated with a solution of Na125I (specific activity 80 GBq/mmol; PerkinElmer, Courtaboeuf, France). The iodination was initiated by adding peracetic acid (1-3%, 100 μL), and quenched 5 minutes later with sodium bisulfite (200 μL, 50 mg/mL). The solution was then neutralized with sodium bicarbonate (200 μL, 50 mg/mL). The product was purified using high-performance liquid chromatography with a semipreparative C-18 RP-column (10 × 250 mm Phenomenex) and a mixture of acetonitrile/0.1 M ammonium acetate (60/40) as mobile phase at a flow rate of 4 mL/min. The retention times of precursor, iodide, and [125I]-CLINDE in this system were 4, 5, and 15 minutes, respectively. The peak corresponding to [125I]- CLINDE was collected, passed through a C-18 Sep-Pack (Waters, Milford, MA), and eluted with ethanol to eliminate the solvent. The final radiolabeled product was assessed using an analytical C-18 RP-column (4.6 × 250 mm Phenomenex), with the same mobile phase and flow rate as described above. The radiochemical yield following purification was about 85%, and the specific activity of [125I]-CLINDE was 80 GBq/mmol.
Animals
All procedures were conducted in accordance with the requirements of the European Community Council for the Care of Laboratory Animals (2010/63/EU) and approved by the local ethical committee (INSERM37-002, agreement n°2012-03-1). Experiments were carried out on adult Wistar rats (Centre d'Elevage René Janvier, Le Genest-St-Isle, France) weighing 282 ± 15 g at the beginning of the experiment. Animals were housed in groups of two per cage in a temperature- and humidity-controlled environment (temperature 22°C ± 1°C; hygrometry 40% ± 7%) under a 12-hour light/dark cycle with food and water available ad libitum.
Surgery
Surgery was performed under isoflurane (AErrane, Baxter, France) anesthesia (4% for anesthesia induction and thereafter 2% for its maintenance, in N2O-O2: 70%-30%). Animals' body temperature (36.9 ± 0.6°C) was monitored with a homoeothermic probe during the whole surgery.
Unilateral striatal QA lesion was induced in 94 adult male Wistar rats as previously described.19,27 This procedure results in a reproducible excitotoxic lesion well delineated to the striatum and ipsilateral cortex. Rats were placed in a stereotaxic David Kopf apparatus (tooth bar: −3.3 mm) and unilaterally injected with 150 nmol of QA (Sigma-Aldrich, Lyon, France) into the right striatum (injection rate: 0.5 μL/min) using a 25 μL microsyringe (Hamilton, Bonaduz, Switzerland) and a micropump (KD Scientific, Holliston, MA). Two microliters of QA were injected at the following coordinates: AP: +0.7 mm, ML: −3 mm, and DV: −5.5 mm from bregma, according to Paxinos and Watson. 28 The injection syringe was left in place for an additional 4 minutes to avoid QA backflow and then slowly removed. The scalp was sutured, and animals were returned to their cages and examined daily until sacrifice.
A complementary set of eight animals (sham) were treated with QA vehicle (0.1 M phosphate-buffered saline [PBS], pH 7.4), according to the same procedure.
Ex Vivo Cerebral Biodistribution of [125I]-CLINDE and Blocking Studies
Ex vivo cerebral biodistribution studies were performed with [125I]-CLINDE in QA-lesioned animals at 1, 4, 7, 14, 30, 60, and 90 days postlesion (dpl) and in sham animals at 14 dpl (n = 6 per group). Rats were intravenously injected (penis vein) with [125I]-CLINDE (0.3-0.4 MBq in 300 μL normal saline). At 14 dpl, nonspecific binding of [125I]- CLINDE was investigated in six other QA-lesioned rats by preinjection, 15 minutes prior to the radiotracer, with an excess (5 mg/kg) of unlabeled PK11195 (the isoquinoline carboxamide TSPO reference ligand, Sigma). Animals were sacrificed 30 minutes post-[125I]-CLINDE injection. Several brain areas (cerebellum, left and right hippocampus, left and right frontal cortex, and left and right striatum) were removed and weighed, and their radioactivity was measured using an automated gamma-counter (Packard Cobra 5010, Packard, GMI, Ramsey, MN). The percentage of injected dose per gram tissue (%ID/g) was calculated by comparison with samples to standard dilutions of the initial dose.
Ex Vivo Autoradiography
Ex vivo autoradiographic studies were performed with [125I]-CLINDE to illustrate the spatial distribution of its cerebral binding in QA-lesioned animals at each time point and in sham animals at 14 dpl (n = 2 per group). At 14 dpl, nonspecific binding of [125I]-CLINDE was investigated in two other QA-lesioned rats by preinjection, 15 minutes prior to the radiotracer, with an excess of PK11195. Animals were intravenously injected (penis vein) with [125I]-CLINDE (2.0 MBq in 300 μL normal saline) and euthanized 30 minutes post-[125I]-CLINDE injection. Brains were removed and frozen at −80°C. Coronal striatal sections (20 μm thickness) at the level of the striatal lesion site, hippocampus, and cerebellum, corresponding to the coordinates AP: 0.72 to 0.60 mm, −4.20 to −4.44 mm, and −13.44 to −13.68 mm, respectively from bregma, 28 were then cut in a cryostat microtome (Jung CM 3000, Leica), thaw-mounted on microscope slides, and air-dried at room temperature. Slides were exposed to iodide-sensitive films (Biomax MRTM Kodak) for 6 weeks and then developed (Kodak L24 revelator), fixed (Kodak 3000 fixator), and dried. Autoradiograms were scanned using a Scanner Epson Perfection 4870 Photo.
Immunohistochemistry Studies
The animals (n = 6 per time point) were euthanzed at 1, 7, 14, 60, and 90 dpl for immunohistochemistry processing. The rats were deeply anesthetized by intraperitoneal injection of pentobarbital (Céva Santé Animale, Paris, France), perfused through the heart with 250 mL of heparinized saline (1 UI/mL; HéparineChoay, Sanofi-Aventis, Vitry-sur-Seine, France), and then followed by 400 mL of 4% paraformaldehyde (PFA; Sigma-Aldrich). The brains were removed and fixed in 4% PFA for 2 hours and then stored for 48 hours in 30% sucrose and frozen at −80°C.
Five free-floating coronal sections, 40 μm thick, of the striatum region were used for immunohistochemistry staining of neurons, activated microglial cells, and astrocytes. Endogenous peroxidase was blocked using 3% H2O2 in 10% methanol and distilled water for 15 minutes. Slices were incubated overnight at room temperature with primary antibodies 1:500 diluted antineuronal nuclei (NeuN, Millipore, Molsheim, France), 1:500 diluted anti-CD11b (Ox-42, AbdSerotec, Düsseldorf, Germany), or 1:1,000 diluted glial fibrillary acidic protein (GFAP; Sigma-Aldrich), 0.1 M PBS supplemented with 0.2% v/v of Tween, and 2% v/v of normal horse serum overnight at room temperature. Sections were then incubated with biotinylated horse antimouse IgG secondary antibodies (AbCys, Paris, France) for 90 minutes at room temperature. Neurons, activated microglia, or astrocytes were visualized by staining with streptavidin-biotin conjugated horseradish peroxidase (AbCys) for 60 minutes at room temperature. Peroxidase was developed for 3 minutes with diaminobenzidine at room temperature. Slices were analyzed under a light binocular microscope (Leica, Westlar, Germany), and histologic analysis was performed with Histolab imagery software (US Histology Laboratories, Rockville, MD). The neurons were visually counted, and the percentage of neuronal loss in the ipsilateral versus the contralateral hemisphere was calculated in matching areas of analysis. For Ox-42 and GFAP, the total surface area occupied by activated microglia or astrocytes was automatically measured by the imagery software and the percentage of increase in the ipsilateral versus the contralateral hemisphere was measured for each section. Five slices per animal, at the level of the striatal lesion site (AP: 0.72 to 0.60 mm from bregma 28 and three areas per hemisphere), were then randomly selected and neurons or surface occupied by activated microglia or astrocytes were counted by two independent operators.
Statistical Analysis
All data are presented as mean ± standard error of the mean. Significance was set at p < .05. Data for multiple variable comparisons were analyzed by one-way analysis of variance (ANOVA) followed by nonparametric analysis (Mann-Whitney and Kruskal-Wallis tests) to determine significant difference. Statistics were performed using Statview software (Abacus Concepts Inc., Piscataway, NJ).
Results
Ex Vivo Longitudinal Follow-up of [125I]-CLINDE Cerebral Distribution and Blocking Studies
The cerebral biodistribution of [125I]-CLINDE at 1, 4, 7, 14, 30, 60, and 90 dpl in rats lesioned with 150 nmol of QA was expressed as the radioactivity levels, in percent injected dose per gram of tissue (%ID/g), and is shown in Figure 1A. At 1 dpl, the accumulation of radioactivity in the right (lesioned) striatum and cortex was significantly increased (p < .05) compared to the contralateral side: 0.36 ± 0.02% ID/g tissue versus 0.16 ± 0.01% ID/g tissue in striatum, that is, +125%; 0.32 ± 0.02% ID/g tissue versus 0.18 ± 0.01% ID/g tissue in cortex, that is, +78%. This difference between both hemispheres was maintained in the striatum up to 60 dpl but was no longer statistically significant at 90 dpl. In cortex, a significant increase in [125I]-CLINDE binding was observed only until 14 dpl.
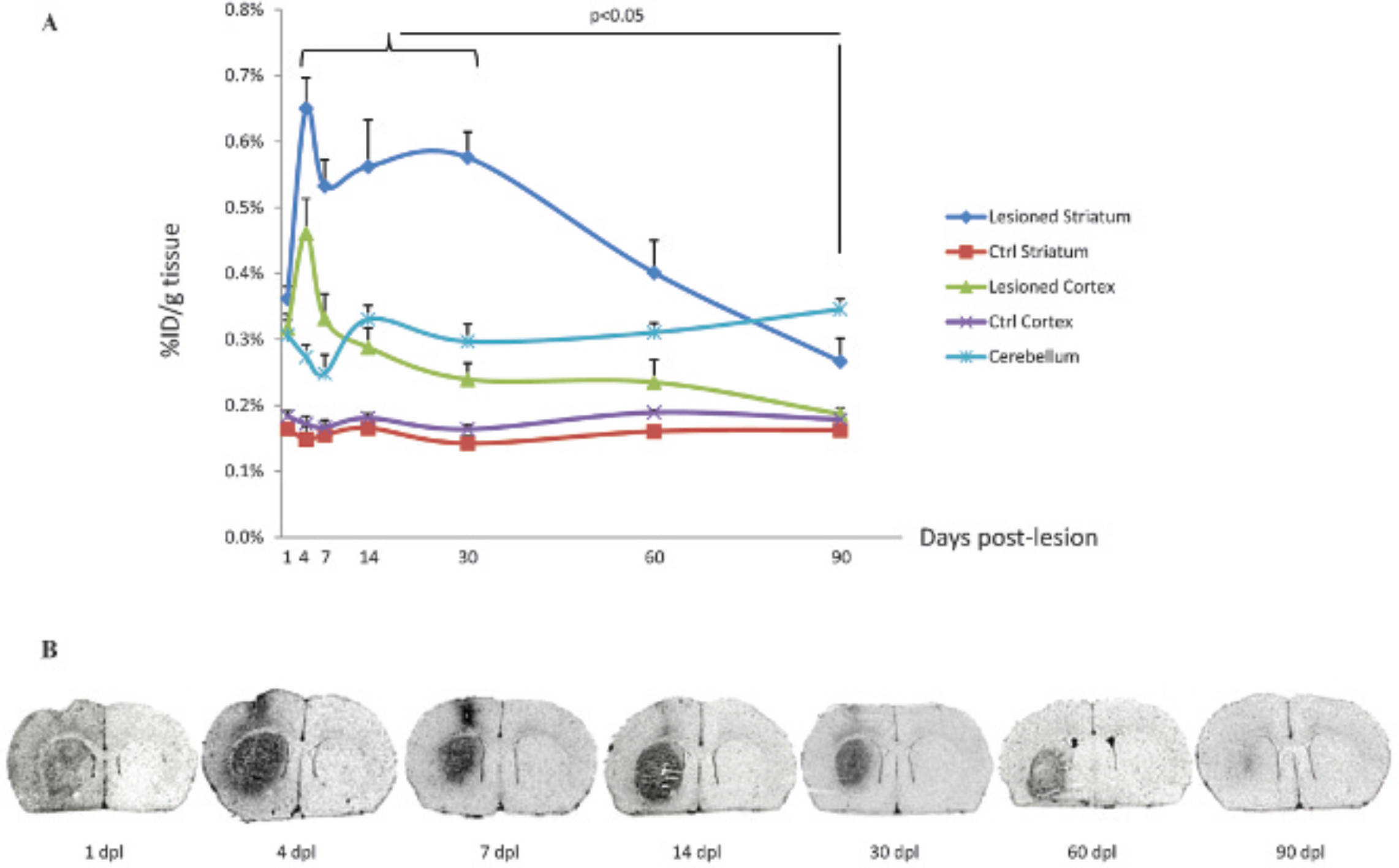
Temporal cerebral characterization of TSPO overexpression after 150 nmol QA unilateral striatal injection using [125I]-CLINDE. A, Brain regional biodistribution (% injected dose/g tissue: mean ± SEM) of [125I]-CLINDE in selected cerebral regions of rats after 1, 4, 7, 14, 30, 60, and 90 days post-QA lesion (dpl), 30 minutes after [125I]-CLINDE intravenous injection. B, Representative ex vivo autoradiographic images showing [125I]-CLINDE accumulation after QA striatal lesion. Coronal sections at the level of striatums from lesioned rats following 1, 4, 7, 14, 30, 60, and 90 dpl are depicted.
In the lesioned striatum, the accumulation of [125I]- CLINDE continued to increase significantly at 4 dpl (0.65% ± 0.05% ID/g tissue, p < .05 vs 1 dpl) and reached a plateau between 4 and 30 dpl. Indeed, we did not observe any statistically significant differences between [125I]- CLINDE uptake at 4, 7, 14, and 30 dpl, whereas all of these time points evidenced a higher value versus 1 dpl (p < .05). After 30 dpl, this value decreased progressively, reaching significance at 90 dpl (0.58 ± 0.04% ID/g tissue at 30 dpl vs 0.27 ± 0.04% ID/g tissue at 90 dpl, p < .05).
In the ipsilateral cortex, the radioactive signal peaked at 4 dpl (0.46 ± 0.05% ID/g tissue, p < .05 vs 1 dpl) and then progressively decreased to return to a basal level, similar to the values observed in the contralateral hemisphere, at 90 dpl.
In the hippocampus, [125I]-CLINDE uptake was low (≈ 0.16 ± 0.02% ID/g tissue) and steady regardless of the brain hemisphere or the considered time point of the longitudinal follow-up.
The cerebellum likewise provides a consistent accumulation of radioactivity whatever the time point, with values between 0.25 ± 0.03% ID/g tissue at 7 dpl and 0.35 ± 0.02% ID/g tissue at 90 dpl, and no significant difference was observed by multiple variable comparison analysis. However, cerebellar [125I]-CLINDE binding appeared significantly higher than the values obtained in the contralateral hemisphere, whatever the considered time point (eg, at 30 dpl: 0.30 ± 0.03% ID/g tissue in cerebellum vs 0.14 ± 0.01% ID/g tissue in striatum contralateral to the QA lesion, p < .05).
Ex vivo autoradiographic images, illustrating the striatal and cortical distribution of radioactivity 30 minutes after [125I]-CLINDE injection to rats lesioned with 150 nmol QA, are presented in Figure 1B. Autoradiographic studies showed preferential localization of [125I]-CLINDE labeling in the ipsilateral striatum and cortex when compared to the contralateral side, with a maximal signal between 4 and 30 dpl. Moreover, at 1 and 4 dpl, the distribution of radioactivity was diffused and homogeneous in the whole ipsilateral hemisphere, whereas it was restricted to the lesioned striatum and the cortical needle track for QA injection at 7, 14, 30, and 60 dpl. At 90 dpl, the increased [125I]-CLINDE ipsilateral binding was almost unnoticeable in comparison with the contralateral side.
Competition studies, consisting of a pretreatment with unlabeled PK11195 (5 mg/kg), 15 minutes before the radiotracer, as well as evaluation of [125I]-CLINDE brain accumulation in sham animals, were undertaken at 14 dpl. The results are presented in Figure 2A.

[125I]-CLINDE binding in QA-lesioned rat brain and sham animals and effects of pretreatment with PK11195 at 14 days after surgery, using ex vivo experiments. A, Brain regional biodistribution (% injected dose/g tissue: mean ± SEM) of [125I]-CLINDE in selected cerebral regions of rats after 1, 4, 7, 14, 30, 60, and 90 days post-QA lesion (dpl), 30 minutes after [125I]-CLINDE intravenous injection. Brain regional biodistribution (% injected dose/g tissue: mean ± SEM) of [125I]-CLINDE and effects of pretreatment with the TSPO-selective ligand PK11195 (5 mg/kg) on [125I]-CLINDE binding in selected cerebral regions of rats lesioned in the right striatum with 150 nmol of QA or injected with its vehicle (sham group), 30 minutes after [125I]-CLINDE intravenous injection. B, Representative ex vivo autoradiographic images showing [125I]- CLINDE accumulation in QA-injected animals, with or without PK11195 pre-injection (competition studies) and in sham animals, 14 days after surgery, at the level of the striatal injection, hippocampus, and cerebellum.
We observed that the accumulation of radioactivity in the ipsilateral striatum was significantly decreased in sham animals compared to the QA-lesioned group at 14 dpl: 0.21 ± 0.01% ID/g tissue for the sham group versus 0.56 ± 0.07% ID/g tissue for the QA group, that is, −62%; p < .05), whereas no significant difference was observed in any other brain region between these two groups. Pretreatment with PK11195 significantly reduced the intensity of ipsilateral striatal and cortical [125I]-CLINDE binding (p < .05), with the same profile of inhibition (decreases of 75% and 69% in striatum and cortex, respectively), whereas no significant difference was observed in the contralateral hemisphere and ipsilateral hippocampus. Moreover, whereas cerebellar radioactivity was similar in the QA-lesioned and sham groups (0.33 ± 0.02% ID/g tissue for both groups), PK11195 pretreatment induced a strong decrease in the signal in cerebellum (0.08 ± 0.01% ID/g tissue, ie, −75%, p < .05).
Ex vivo autoradiographic images, comparing the striatal, hippocampal, and cerebellar localization of radioactivity 30 minutes after [125I]-CLINDE injection between sham, PK11195-preinjected, and QA-lesioned animals at 14 dpl, are presented in Figure 2B. Autoradiographic studies evidenced that pretreatment with PK11195 markedly decreased striatal [125I]-CLINDE binding and that in the sham group, accumulation of radioactivity spatially corresponded to the needle track for vehicle injection. Moreover, it confirmed that [125I]-CLINDE cerebellar distribution was similar in sham and QA-lesioned animals and inhibited by PK11195 pretreatment.
Immunohistochemistry Studies
A time course of neuronal loss, as well as activation of two glial subpopulations, microglia, and astrocytes, has been analyzed by immunohistochemistry in contralateral and ipsilateral striatum of rats at days 1, 7, 14, 60, and 90 after QA injection (Figure 3).

Immunohistochemistry of NeuN, Ox-42, and GFAP in rat striatum. A, B, and C represent the same areas of contra- and ipsilateral striatum (1 and 2, respectively) at 1, 14, and 90 days after surgery. Magnification was X20. Data are means ± SEM. Bars: 20 μm. A, NeuN. Neurons staining in contra- and ipsilateral striatum (1 and 2, respectively) at 1, 14, and 90 days after surgery. Data for each time are expressed as relative neuronal loss in the ipsilateral hemisphere versus the contralateral hemisphere (3). B, Ox-42. Activated microglia immunochemistry in contra- and ipsilateral striatum (1 and 2, respectively) at 1, 14, and 90 days after surgery. Data for each time are expressed as relative increasing of microglia activation in the ipsilateral versus the contralateral hemisphere (3). C, GFAP. Activated astrocytes staining in contralateral and ipsilateral striatum (1 and 2, respectively) at 1, 14, and 90 days after surgery. Data for each time are expressed as relative increasing of astrocytes activation in the ipsilateral versus the contralateral hemisphere (3).
The excitotoxic QA lesion significantly increased (p < .05) OX-42 and GFAP immunoreactivity with respect to the nonlesioned side of the brain as early as 1 dpl, that is, concomitantly with significant evidence of neuronal loss.
Neuronal loss was significantly higher at day 14 (83.9 ± 2.2%) with respect to the other time points (p < .05). Neuronal loss decreased with time after QA injection and was 12.5 ± 3.4 at day 90.
Microglia immunostaining using Ox-42 showed cells with the ramified morphology of reactive microglia in the lesion and evidenced an increase in CD11b immunoreactivity between days 1 and 14, where microglia activation was the highest (4443.9 ± 1030.7%). Beyond this time point, immunolabeling showed a clear decrease in microglial expression in ipsilateral striatum to 257.3 ± 89.6% in relation to the control at 90 dpl. Measurement of GFAP immunoreactivity demonstrated that QA injection increased astrogliosis relative to the nonlesioned control at days 7 and 14 (760.9 ± 63.4% and 704.6 ± 57.4%, respectively) and decreased further after (p < .05).
At 90 dpl, immunoreactivity of OX-42 and GFAP in ipsilateral striatum remained slightly but significantly higher when compared to the contralateral hemisphere; likewise, neuronal density in ipsilateral striatum remained lower than that measured in the contralateral hemisphere.
Discussion
Expression of TSPO is dramatically upregulated under neuropathologic conditions, including neurodegenerative diseases, brain trauma, and cerebral ischemia. This overexpression of TSPO has been related to the activation of glial cells after brain injury. Thereby, TSPO is considered an attractive and sensitive marker for the quantification and visualization of neuropathologic changes induced during cerebral inflammation. We showed in a previous article that the TSPO single-photon computed tomography (SPECT) ligand CLINDE accurately reflects the changing degree of neuroinflammation in the same rat model of excitotoxic lesion induced by striatal lesion with QA. 19 We demonstrated that the large TSPO upregulation in the lesioned brain hemisphere was correlated with the intensity of the lesion and that in vivo accumulation of CLINDE was able to discriminate these various degrees of microglial activation.
In the present study, we evaluated over a long time period (1 to 90 dpl) the time-dependent evolution in [125I]-CLINDE signal induced by a 150 nmol striatal injection of QA, compared to that in the contralateral side. In ipsilateral striatum, ex vivo cerebral biodistribution, a method recognized to reflect the extensive distribution of the tracer in living animals, evidenced an increased binding of [125I]-CLINDE from 1 day after striatal lesion, reaching a maximal value at day 4, followed by a plateau up to 30 dpl, and before a progressive return to normal values at 90 days, that is, similar binding on the lesioned and intact sides.
The binding profile in the ipsilateral cortex was different, showing a progressive increase beginning at 1 dpl and peaking at 4 dpl, with a 170% increase on the lesioned compared to the intact side, followed by a progressive decrease until a basal level at 90 dpl, as in the striatum. This discrepancy correlates with a widespread breakdown of the blood-brain barrier (BBB). Indeed, this QA model has the drawback of causing a rupture of the BBB during injection, which may influence brain distribution of the tracer. This BBB disruption is reversible, and integrity is restored about 7 days after surgery. 29 Therefore, cortical peak at 4 dpl might be related to this BBB transitory breakdown, and the lasting cortical uptake probably reflected the presence of a mechanical lesion due to the needle track, whereas microglial activation was still present in the excitotoxic striatal lesion. Accordingly, ex vivo autoradiographic imaging confirmed the trend of a higher uptake of [125I]-CLINDE associated with neuroinflammation in both ipsilateral striatum and cortex. This BBB disruption was also evidenced by the diffused and homogeneous distribution of radioactivity in the whole ipsilateral hemisphere in autoradiography at 1 and 4 dpl.
The inflammatory astroglial and microglial reactions, in parallel to neuronal loss, were also characterized by immunohistochemistry at 1, 7, 14, 60, and 90 days after QA injection. The results showed an overall increase in glial expression in the lesioned area and indicated that this increase was maximal at 7 dpl for astrocytes and at 14 dpl for microglia. Indeed, the number of CD11b-reactive cells (ie, of monocytic lineage) increased progressively up to day 14 and decreased in the following weeks, until day 90. The origin of these OX-42-positive cells can be the activation of central resident microglia or the migration within brain parenchyma of peripheral circulating leukocytes after BBB disruption.
Immunohistochemistry using a primary antibody targeting CD11b antigen as a marker of microglia was used in previous studies to characterize the temporal profile of microglial activation in a slightly different model of excitotoxicity in which QA was administered at a higher dose (210 nmol vs 150 nmol in the present work) and further forward in the striatum (AP: +1.5 mm vs +0.7 mm in the present work 23 ). The increase in activated microglia expression was detected in the striatum at 8 dpl, was still present at 30 dpl, and then disappeared at 60 dpl. Therefore, this time course of microglial activation closely paralleled the pattern of uptake of [125I]-CLINDE we described here.
The expression of GFAP-immunoreactive astrocytes peaked at 7 and 14 dpl, in agreement with the results observed by Ryu and colleagues in a close model of QA striatal injection. 30 This observation is consistent with reports of increased microglial populations at 7 days after ethanol-induced neuronal insults, followed by a later decrease at 30 days. 31
Interestingly, the time course of [125I]-CLINDE binding that we observed using ex vivo biodistribution and autoradiography matched with the temporal profile of both microglia activation and astrogliosis. This observation is not in agreement with data from Moresco and colleagues, suggesting that OX-42 signal decreased after 30 dpl, whereas [11C]-PK11195 binding still significantly enhanced at 60 dpl. 23 The authors suggested that this discrepancy could be explained by the TSPO expression in delayed reactive gliosis, but this hypothesis was not demonstrated using GFAP-specific antibody. Nevertheless, these data were based on a low number of animals (n = 1 for OX-42 and n = 3 for [11C]-PK11195 versus n = 6 for both immunohistochemistry and CLINDE studies in our experiments). Moreover, the relevance of our finding is also based on the fact that analyses were extended to later time points up to 90 days after lesion, and both molecular imaging ([125I]-CLINDE) and glial immunoreactivity (OX-42 and GFAP) demonstrated the transitory process of neuroinflammation in this model.
The relative contribution of microglia and astrocytes in TSPO overexpression in the central nervous system during neuroinflammation is a subject of debate 32 and may also depend on the animal model, species, and strain used and on the time course of the neuroinflammatory process. To clarify this point, we used combined autoradiographic and immunohistochemical experiments, using OX-42 as a marker of microglia, GFAP for astrocytes, and [125I]- CLINDE for TSPO. It suggests that TSPO overexpression was the consequence of both microglial investigation and astrogliosis because the plateau observed in CLINDE striatal binding from 7 to 14 dpl was temporally correlated to the successive peaks of GFAP and OX-42 immunoreactivity. Moreover, ex vivo autoradiography evidenced the striatal colocalization of [125I]-CLINDE binding with both antibodies immunostaining. This cellular expression of TSPO shall be confirmed using specific anti-TSPO antibodies in immunocostaining with OX-42 and GFAP because we did not achieve this point in the present study for technical reasons.
In our model, neurons staining using NeuN antibody showed an important neuronal loss (about 85% at 14 dpl), although it progressively decreased thereafter, to practically disappear at 90 dpl. Similar results have been previously described after intrastriatal QA injection, but these studies did not explore this “neurogenesis-like” process beyond 6 weeks after surgery.23,33–35 The mechanisms involved in this neuronal regeneration could be linked to the migration of newborns neurons in the striatum. Indeed, Tattersfield and colleagues evidenced an early increase in progenitor cells in the subventricular zone (SVZ) following QA injection. 35 More recently, another study in a QA model showed that these cells expressing the doublecortin marker for neuroblasts migrate from the SVZ into striatum during the first days after injury. 34 Collin and colleagues observed the same cell proliferation in the SVZ following QA injection, related to an important increase in migrating neurons in the ipsilateral striatum at 14 dpl. 33 They also showed that these “newborn cells” became NeuN positive, a marker of neuronal maturation, at 6 weeks after surgery. Moreover, the level of this neuronal proliferation was correlated with the amount of QA injected. Therefore, the neuronal regeneration we observed in the present work might be explained by this neurogenesis process, demonstrated in these different studies using QA.
This study highlighted the comparative time courses of neuroinflammation and neurodegeneration in this excitotoxic model, showing the concomitance and transitory nature of both neuronal loss and reactive gliosis processes in the lesioned striatum. Therefore, this finding provides the possibility to propose a time window during which the administration of factors able to block or reduce the neuroinflammation process could also be able to block or slow down the degenerative process, at least in this animal model. Thus, the beneficial effects of different agents able to act on microglial activation have recently been reported,9,30 and it is of great interest for therapy development, follow-up of the disease, and global understanding of pathogenesis to also consider their effectiveness longitudinally. In this regard, such a therapeutic approach could be accessed in a clinical setting through in vivo molecular imaging methods that can monitor concomitantly neuroinflammation and disease progression and assess noninvasively the efficacy of potential therapeutics targeted to diminish neuroinflammation.
Furthermore, blocking studies strongly supported the view that this increase in [125I]-CLINDE uptake was specific binding to TSPO because it was displaced by pretreatment with a saturating dose of cold TSPO-specific ligand PK11195. Pretreatment with PK11195 reduced the striatal radioactive signal by 75%, which was similar to the inhibition measured with PK11195 in the same excitotoxic animal model, 19 confirming equilibrium in TSPO expression. In this respect, our present results support the use of the contralateral region rather than cerebellum to optimize the target to background ratio measurement, at least in animal models induced by a focal cerebral lesion. Indeed, an increase in CLINDE uptake evidenced a well-localized area rich in TSPO, whereas the brain's other regions remain devoid of TSPO, with the exception of the cerebellum. Remarkably, CLINDE uptake was significantly increased in the cerebellum compared to the left hippocampus as the control for each time point of our longitudinal follow-up. At 14 dpl, the target to background ratio between lesioned striatum as target and contralateral striatum or cerebellum as reference region was 3.3 and 1.7, respectively. Interestingly, we observed a ratio with cerebellum as a reference region at 7 dpl of 2.1, similar to data obtained by our group in a previous study in the same model. 19 This elevated CLINDE cerebellar uptake was not related to any induced neuroinflammatory challenge because it was present in sham animals (0.33% ID/g tissue at 14 dpl, identical to the QA-lesioned group) but was strongly associated with TSPO expression because it was markedly inhibited by PK11195 pretreatment (0.08% ID/g tissue at 14 dpl, ie, −75%). Autoradiography confirms this constitutive presence of TSPO in layers of the cerebellum. Several authors previously documented the fact that cerebellum in rodents is a TSPO-rich brain region36,37; nevertheless, it is still used as a reference region for quantification of radioactive signal in molecular imaging studies using TSPO radioligands,23,38 which may represent a limitation of these studies.
Conclusion
This work highlights that molecular imaging techniques using selected radiopharmaceuticals combined with immunohistochemistry provide complementary information to follow disease progression in a preclinical model of Huntington disease. The temporal behavior of neuroinflammation evidenced a transitory process, characterized by an elevated and stable TSPO expression from 4 to 30 days after QA administration, evaluating by microglial activation and astrogliosis in parallel with neuronal death measurement. These results suggest that there might be a therapeutic window for antiinflammatory agents in the early stage of the disease and will be very useful for the longitudinal evaluation of the potential effectiveness of novel neuroprotective strategies over a long time period.
Footnotes
Acknowledgment
Financial disclosure of authors and reviewers: None reported.