
Abstract
In heart valve tissue engineering, assessment of cell migration under dynamic states can provide insights on the evolving tissue structure. We labeled human vascular smooth muscle (SMCs), endothelial (ECs), and bone marrow–derived mesenchymal stem cells (BMSCs) with superparamagnetic iron oxide (SPIO) microparticles and visualized them using magnetic resonance imaging (MRI) under steady flow. We determined that vascular cells were able to remain reasonably viable and proliferate well after being labeled with SPIO microparticles (200 μg/mL) for 48 hours. SPIO-labeled cells were successfully visualized using T2* contrast. When physiologically representative shear stresses (5–6 dynes/cm2) were applied to SMC-EC coculture–seeded scaffolds, hypointense regions seemed to have decreased after 2 weeks in some locations, whereas others revealed sustained levels of T2* contrast; similar observations were seen in the case of BMSC-seeded scaffolds. This could be attributable to increased out-of-plane cell migratory activity, which occurred from the fluid-induced mechanical cues received, which was not previously evidenced in static culture. Vascular cells and BMSCs were labeled with remarkably high concentrations of SPIO. Moreover, steady fluid flow enhanced intrascaffold cell migration of vascular SMCs and ECs as well as BMSCs, which, in turn, significantly improved construct cellularity and extracellular collagen content
HEART VALVE ANOMALIES pose serious health concerns, often requiring valve replacements. Available valve replacements in the market are mechanical, xenograft fixed-tissue, and homograft valves. However, current prosthetic heart valves have several limitations. Mechanical valves are prone to clot formation; therefore, lifelong anticoagulant therapy is required. 1 Xenograft fixed tissues, commonly referred to as bioprosthetic heart valves, do not require anticoagulants but have limited durability and are prone to calcification. 2 Homograft valves are limited in availability and like bioprosthetic valves may calcify and undergo premature, structural breakdown. 3 Given that many patients are children, repetitive replacement surgeries are required because available prosthetic valves do not accommodate somatic growth.4–6 Tissue-engineered heart valves (TEHVs) could potentially address these limitations, providing for growth, self-repair, infection resistance, and a permanent approach for replacing anomalous valves.
The generic approach for the creation of TEHVs uses a bioabsorbable scaffolding material, seeded with cells and grown in vitro, to ultimately produce three-dimensional (3D) extracellular matrix (ECM) resembling native valve morphology. 7 During the culture process, cells organize, grow, proliferate, and deposit engineered ECM on the scaffold. The results in the TEHV arena appear to be promising: for example, Sutherland and colleagues constructed TEHVs by seeding bone marrow–derived mesenchymal stem cells (BMSCs) onto nonwoven scaffolds that functioned in the pulmonary outflow tract of sheep for 8 months. 7 More recently, several studies have demonstrated the need to mechanically condition engineered heart valve tissues in vitro so that the constructs will possess sufficient biologic, biochemical, and mechanical properties to be able to continue tissue remodeling activity and withstand hemodynamic forces in vivo. Indeed, mechanical stimulation of engineered tissues grown in bioreactor devices has been shown to improve the physical integrity of the constructs.8–10 The mechanical environment would potentially include physiologic flows for conditioning the developing valve tissues in a sterile setting prior to implantation.10–12 However, tissue remodeling during initial phases after implantation will depend on cell migratory events to and from the scaffold and will involve both the cells used to seed the scaffolds and the endogenous cells of the artery. Thus, it is clear that the ability to noninvasively and nondestructively track the fate of the cells is critical in assessing in vitro mechanical conditioning effects on cell distribution and subsequent engineered ECM formation.
Relevant cell sources pertinent in TEHV efforts include human vascular cells (native artery smooth muscle [SMCs], endothelial cells [ECs], and stem cells. Vascular cells (SMCs and ECs) have been shown to promote heart valve tissue formation.4,9,13 Hoerstrup and colleagues and Mol and colleagues observed enhanced tissue formation from vascular cells in mechanically conditioned leaflets.9,13 They reported significant net collagen and deoxyribonucleic acid (DNA) content in their engineered leaflets over static controls. On the other hand, from a stem cell perspective, studies using BMSCs have shown that after TEHV implantation, the BMSCs exhibited sufficient pluripotency for potential vascularization of the engineered tissues. 7
For monitoring of cells, a cell-tracking agent is usually necessary. Such an agent has to be safe to use; that is, it does not alter cell activity and should permit retention within the cell for a reasonable period to allow for visualization at longitudinal time points. 14 One approach would be to label the cells with superparamagnetic iron oxide (SPIO) microparticles and visualize them as regions of signal voids by magnetic resonance imaging (MRI). Other cell-tracking methods include labeling with green fluorescent protein; however, this method is harmful for the tissue as it may interfere with the protein synthesis of the cells and does not provide sufficient penetration or absorption to be able to detect deep tissues, which is needed for the heart valve application. 15 On the other hand, SPIO microparticles have been shown to work in conjunction with MRI monitoring without harming the cells.16–19 For example, Terrovitis and colleagues showed that labeling BMSCs with ferumoxides was not toxic to the cells and that the cells could further be visualized in collagen gels or scaffolds. 19 Ko and colleagues showed that cellular MRI methodologies could be used to visualize an implanted scaffold and the cellular behavior within scaffolds implanted in mice. 20
MRI has been reported to be useful in the detection and tracking of SPIO-labeled cell fate in vivo. 17 MRI generally allows SPIO-labeled cell detection by accelerated T2* decay, which will present as hypointense regions caused by the intracellular iron oxide particles. 21 This visualization is necessary from the beginning of the in vitro culturing period of the engineered construct and could in theory be progressively tracked, even after implantation. 7
Previous findings in our laboratory showed that the spatial positions of seeded SPIO-labeled SMCs and ECs in 3D tissue-engineered constructs cultured under static conditions were relatively unchanged over a period of 12 days. 21 As an extension of this work, here, 3D cellular scaffold constructs conditioned with the steady flow of culture media were visualized and monitored by MRI to assess the effects of the fluid flow environment on longitudinal changes in intrascaffold cellular distribution. The motivation for this work is connected to the underlying premise for heart valve tissue engineering protocols, that mechanical in vitro conditioning is necessary for proper construct development. Therefore, assessment of temporal cell migratory patterns under these mechanical environments would provide tremendous insights on the evolving engineered tissue structure, in a noninvasive manner using MRI. We also investigated a much higher concentration of SPIO microparticles compared to our previous work (200 μg/mL versus 13.3 μg/mL used earlier 21 ) to determine the extent to which intracellular iron oxide, T2* MRI contrast could be maximized without harming the cells.
Materials and Methods
Two-Dimensional Studies
Cell Labeling
Various concentrations of the superparamagnetic polymer microspheres, or “Bangs particles” (particle size ≈ 0.86 μm, Bangs Laboratories, Fishers, IN), were used to label the vascular cells (human pulmonary artery–derived SMCs and ECs, Genlantis, San Diego, CA), at two different incubation times (24 and 48 hours). The Bangs particles consisted of a magnetite core encapsulated with a styrene/divinyl benzene polymer. Simultaneously, the particles contained a red fluorescent dye (660 nm excitation, 690 nm emission).
After coculture expansion (SMCs and ECs), ≈ 3 million cells were replated equally into 15 chamber slides (Fisher Scientific, Pittsburgh, PA). Vascular cells were labeled for a 24-hour period with three different concentrations of SPIO microparticles (0.23 g of Fe per milliliter of solution: 50, 100, and 200 μg/mL). Concomitantly, during the same 24-hour period, cells were exposed to 4.5 μg/mL of a transfection agent (protamine sulfate, Sigma-Aldrich, St Louis, MO). A second group of vascular cells was labeled with ferumoxides in the same manner except that the incubation time was doubled, that is, 48 hours. Finally, BMSCs (Science Cell Research Laboratories, Carlsbad, CA) were labeled with SPIO microparticles at a concentration of 200 μg/mL for 24 hours while being exposed to 12 μg/mL of a transfection agent (protamine sulfate) following the protocol used by Suzuki and colleagues. 22 It is important to point out that after the labeling durations of 24 and 48 hours, cells were washed several times using phosphate-buffered saline (PBS) to remove exogenous iron oxide. SPIO-labeled cells were then isolated using a magnetic sorter (Capture-Tec Stand, Invitrogen, Carlsbad, CA). Only strongly labeled cells separated from the cell suspension and migrated toward the magnetic stem of the sorting device. After about 10 minutes, a dark pellet, of cells indicative of robust intracellular SPIO uptake, was found to aggregate adjacent to the magnetic stem (visual observations; data not provided). Only these cells were used for further experimentation to ensure that the cells were adequately labeled with the Bangs particles. Note that unless otherwise stated, subsequent experiments with the SPIO-labeled SMC-EC cells (for proliferation, viability, histology, fluorescence microscopy, scaffold seeding, MRI) used an SPIO concentration of 200 μg/mL with a 48-hour incubation time for reasons explained in the Results section. In addition, all subsequent reporting of the time scale of SPIO-related experiments included this 48-hour SPIO incubation period (eg, 1-week proliferation assessment refers to 48 hours of SPIO incubation followed by 1 week of cell proliferation).
Proliferation and Viability
SPIO-labeled and unlabeled cells (SMCs and ECs) were cultured for 1 week to assess proliferation and viability. Cells at day 0 were plated in T12.5 cm2 flasks (Fisher Scientific) with live cells in the order of 240,000 to 270,000 cells/flask. Five flasks were prepared for each of the four iron oxide concentrations being evaluated (0, 50, 100, and 200 μg/mL). SPIO-labeled cells underwent iron oxide incubation (as described in the Cell Labeling section) for 48 hours prior to the 1-week culture period. ECs were cultured using proprietary culture medium from the manufacturer (Genlantis), whereas SMCs were grown in vitro using Dulbecco's Modified Eagle's Medium (Lonza, Basel, Switzerland) with 4.5 g/L glucose and L-glutamine (Invitrogen) supplemented with 10% fetal bovine serum (ATCC, Manasas, VA), and 1% penicillin/streptomycin (Invitrogen). At both the beginning and end of a 1-week period, live and dead cells were distinguished from each other using a trypan blue exclusion assay (Sigma-Aldrich, St. Louis, MO) and subsequently counted using a hemocytometer.
Prussian Blue Histology
After culturing SPIO-labeled cells (SMCs and ECs) for 1 week, they were rinsed with PBS and detached from the T-75 cm2 flask (Fisher Scientific) by 0.25% trypsin–ethylendiamine tetraacetic acid (EDTA) (ATCC). Cell pellets were collected in a 15 mL conical tube (Fisher Scientific) and transferred to chamber slides (n = 6 per cell type). After the cells attached overnight, they were fixed with 10% formalin for further detection of intracellular iron oxide. Slides (n = 3 per cell type) were then incubated in a 1:1 working solution of 2% potassium ferrocyanide and 2% hydrochloric acid for 20 minutes at 55°C and washed in tap water followed by a counterstain of nuclear fast red for 5 minutes. The slides were then dehydrated through alcohol and cleared in xylene. After mounting the slides with Permount (Fisher Scientific), they were examined under an upright microscope for the presence of iron particles inside the cells.
Fluorescence Microscopy
After 1 week of culture, slides (n = 3 per cell type) were counterstained for filamentous-actin (F-actin, Invitrogen) using Alexa Fluor 488–conjugated phallotoxin at 5 U/mL for 20 minutes at 26°C and washed in PBS. The slides were then mounted and stained for DAPI with Vectashield Mounting Media (Invitrogen) and examined under an inverted fluorescence microscope (Nikon Eclipse Ti, Nikon Instruments Inc., Melville, NY) for the presence of the red tag contained in the magnetic core of the Bangs particles (660 excitation, 690 emission).
MRI of Cells in Two-Dimensional Culture
MRI data sets were collected on a 4.7 T (200 MHz) 40 cm bore magnet with a Bruker Avance console (Bruker Biospin MRI GmbH, Ettlingen, Germany) using an actively shielded gradient set. To increase the sensitivity of signal detection, a linear birdcage radiofrequency (RF) coil with a diameter of 35 mm was used for imaging the SPIO-labeled SMCs and ECs that were plated on individual chamber slides (Fisher Scientific). A two-dimensional (2D) gradient echo fast imaging pulse sequence with 180 μm resolution and 10 ms echo time (TE) was employed to visualize the SPIO-labeled cells in the chamber slide.
3D Studies
Cell Seeding
A nonwoven 50:50 blend of polyglycolic acid (PGA) and poly-L-lactic acid (PLLA) scaffold (Biofelt, Concordia Fiber, RI) was used for cell seeding. This scaffold has an approximate fiber diameter of 0.012 to 0.015 mm and a density of 60 mg/cc. Its benefits include its high porosity factor (> 97%). The PGA/PLLA scaffold has already successfully demonstrated its ability to rapidly grow cells and form an organized 3D tissue structure.1,5 Rectangular scaffold samples (n = 12) of dimensions (≈ 17 mm × 6 mm × 1.5 mm) were then ethylene oxide gas sterilized (AN 306, Anprolene, Andersen Products Inc., Haw River, NC) for 12 hours. Next, each sample was placed in a hybridization tube and seeded with SPIO-labeled SMCs (≈ 87% of total cell population) and SPIO-labeled ECs (≈ 13% of the total cell population) at a density of 2 × 106 million cells/cm2. The hybridization tubes were subsequently placed on a rotisserie (Fisher Scientific) and rotated at 8 rpm inside a standard cell culture incubator operating at 37°C and 5% CO2 for a period of 8 days. In vitro culture duration for all SPIO-labeled, SMC-EC-seeded scaffold samples consisted of 8 days of static conditioning in this manner followed by an additional 14 days of continued static culture for six of the samples and 14 days of dynamic conditioning for the remaining six samples.
For dynamic conditioning, SPIO-labeled, SMC-EC-seeded scaffolds were secured in a flow chamber of a custom-built, MRI-compatible bioreactor (Figure 1A) previously used for cell-tracking studies.12,21 In brief, the bioreactor is capable of replicating physiologic hemodynamic conditions during in vitro tissue development, which is believed to be an essential factor in regulation of engineered heart valve tissue formation.5,10,13 The 3D specimens were cultured under dynamic conditions, specifically with continuous exposure to shear stresses on the order of 5 to 6 dynes/cm2,12,23,24 using a steady flow rate of 850 mL/min. Subsequently, unlabeled SMC-EC cellular scaffolds also underwent the same static culture and bioreactor experiments as the SPIO-labeled counterparts, except that the unlabeled constructs were subjected to DNA and biochemical content assessment (rather than MRI) at the conclusion of the 22-day culture period. Finally, the same experimental protocol was also used to condition three SPIO-labeled BMSC scaffolds in the bioreactor under identical dynamic steady shear stress (5–6 dynes/cm2) environments (using the same cell culture media components as described earlier for SMCs).
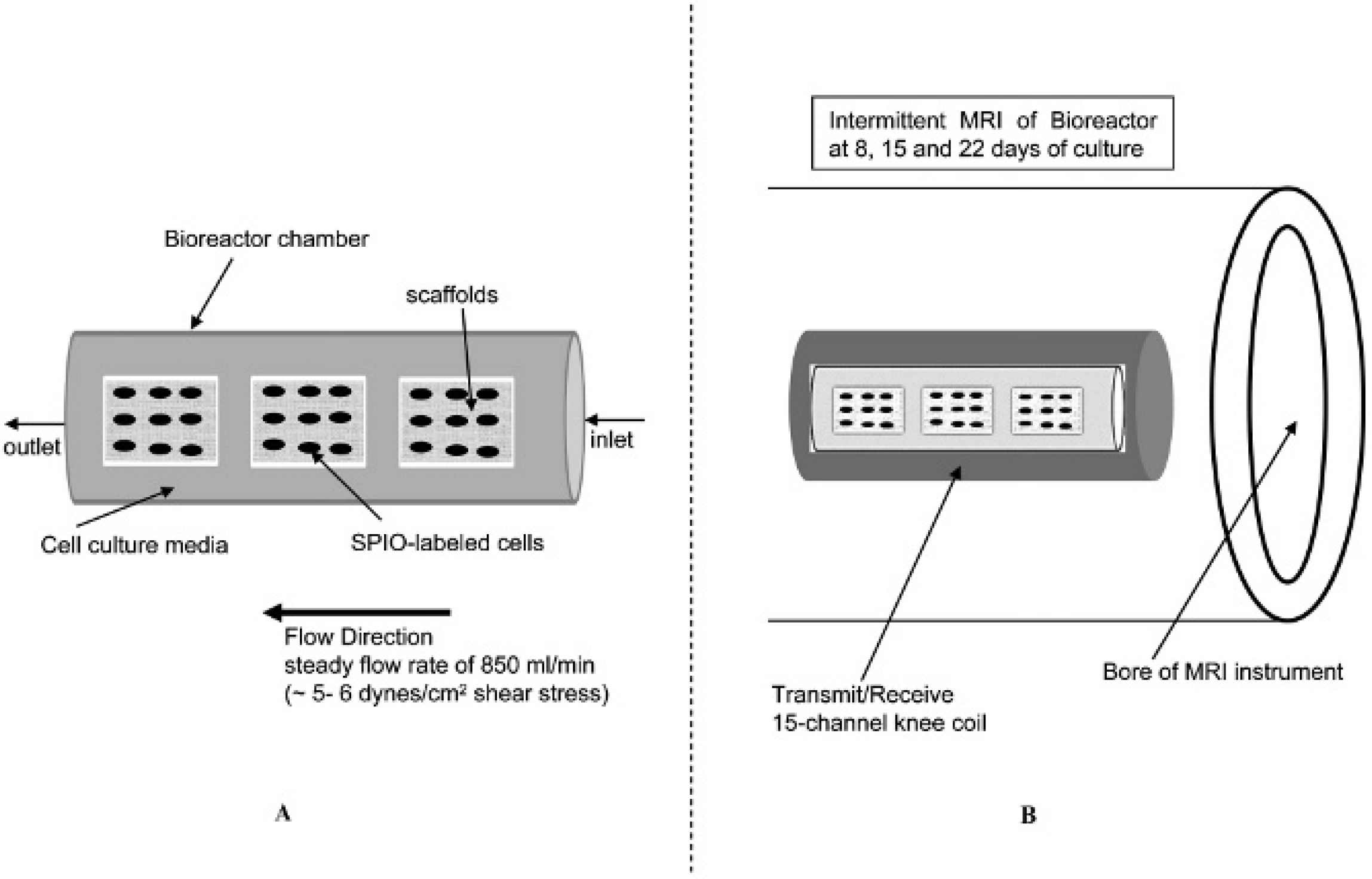
A, Bioreactor flow chamber. B, Illustration of sample placement for MRI experiments.
MRI of Cells in 3D Culture
MRI data sets of the samples housed within the bioreactor flow chamber were collected from a 3.0 T (123 MHz) clinical field, 60 cm bore magnet with a Magnetom Trio, (Tim, Siemens, Erlangen, Germany) console using an actively shielded gradient set. MRI was conducted at intermittent time points during the 22-day culture process, whereby flow was temporarily arrested and the bioreactor flow chamber was placed within the MRI instrument (Figure 1B); this occurred after 8, 15, and 22 days of steady flow mechanical conditioning. A transmit/receive 15-channel knee coil was used to encapsulate the bioreactor chamber. A 3D gradient echo pulse sequence (flash 3D with RF spoiling) was employed. The RF spoiled flash 3D data set was collected, employing a matrix of 192 × 192 × 72 pixels and a field of view of 60 mm × 60 mm × 22 mm, resulting in an isotropic resolution of ≈ 310 μm. A TE of 11.2 ms and a relaxation time (TR) of 100 ms were used to acquire images to visualize the SPIO-labeled SMC-EC-seeded scaffolds (labeled with 200 μm SPIO microparticles for 48 hours) inside the bioreactor. SPIO-labeled, BMSC-seeded scaffolds were then visualized with MRI in an identical manner compared to the vascular cells.
DNA Quantification
At the end of 22 days of culture, the DNA content of the unlabeled SMC-EC-seeded scaffolds from the static and dynamic (steady flow) groups was quantified by a technique adapted from Ramaswamy and colleagues. 23 For each assay, thin samples (8 × 7 × 1 mm) were cut from SMC-EC-seeded scaffolds along their long axis and weighed prior to extraction. Each sample was then placed in a microcentrifuge tube and extracted in 1 mL of 0.125 mg/mL papain solution for 10 hours in a 60°C water bath. The 0.125 mg/mL papain solution was made prior to use by adding L-cysteine dihydrochloride (Sigma-Aldrich) to phosphate-buffered EDTA (PBE; Sigma-Aldrich, to yield a concentration of 10 mM, and subsequently by adding papain (10 U/mg (P4762); Sigma-Aldrich) to arrive at a final concentration of 0.125 mg/mL. The PBE solution was made by adding sodium phosphate dibasic (Sigma-Aldrich) and EDTA (Sigma-Aldrich) to deionized water at concentrations of 100 and 10 mM, respectively. The PBE solution was balanced to a pH of 6.5 with 0.5 N hydrochloric acid (Sigma-Aldrich) and sterile filtered using a vacuum filtration unit (0.2 mm polyethersulfone membrane, Nalge Nunc International Corporation, Rochester, NY). The extracts were assayed using the PicoGreen double-stranded DNA quantitation kit (Molecular Probes, Eugene, OR) as per the manufacturer's instructions and a microplate reader (Synergy HT, Biotek, Winooski, VT).
ECM Quantification
At the end of 22 days of culture, the ECM biochemical content of the unlabeled SMC-EC-seeded scaffolds from the static and dynamic (steady flow) groups was assessed. Collagen and sulfated glycosaminoglycans (S-GAGs) were assayed following a similar technique used by Ramaswamy and colleagues. 23 In brief, for the collagen assay, thin samples (≈ 8 × 7 × 1.5 mm) were cut from SMC-EC-seeded scaffolds along their long axis and their wet weights were measured prior to extraction. Total collagen was extracted from samples using a solution of 0.5M acetic acid (Sigma-Aldrich) and pepsin (1 mg/ml; Sigma-Aldrich). Each sample was placed in a microcentrifuge tube and incubated in 1 mL of extraction solution on an orbital rocker (Orbitron Rotator, Boekel Scientific, Feasterville, PA) at 4°C for 16 hours. For the S-GAG assay, the same extract used for the DNA assay was used. Following the extraction procedure, the collagen and S-GAG extracts were assayed by first reading absorbances of standard solutions and creating a calibration curve and subsequently converting microplate reader-measured absorbances (Synergy HT, Biotek) to normalized biochemical mass content (μg/g wet weight). The specific steps of the assays were adhered to and are described in the manufacturer's protocols (Sircol and Blyscan assay kits for collagen and glycosaminglycan [GAG], respectively, Biocolor Ltd. Newtownabbey, Northern Ireland) using a microplate reader (Synergy HT).
Statistical Analysis
Statistical analysis by way of independent t-tests was performed (SPSS version 16.0, IBM, Armonk, NY) to evaluate significant differences in ECM and DNA quantification results (n = 3, replicates = 5) between control and experimental groups, that is, static (no flow) versus steady flow culture conditions. A value of p < .05 was interpreted to mean that the differences found were significant.
Results
Viability and Proliferation
One-week viability and proliferation of the vascular cells (SMCs and ECs) unlabeled (0 μg/mL SPIO concentration) and labeled at 50, 100, and 200 μg/mL of iron oxide were assessed following 48 hours of the iron oxide incubation period. The percentage of cell viability was computed as follows: (number of live cells/(number of live cells + dead cells) * 100%). The mean percentage of cell viability (n = 5 samples/group) after 1 week of culture was found to be as follows: SMCs: 89.2% (0 μg/mL SPIO), 93.9% (50 μg/mL SPIO), 95.3% (100 μg/mL SPIO), and 74.8% (200 μg/mL SPIO); ECs: 95.3%(0 μg/mL SPIO), 94.8%(50 μg/mL SPIO), 86.8% (100 μg/mL SPIO), and 87.9% (200 μg/mL SPIO).
The percentage of cell proliferation was computed as follows: (number of live cells at day 7 – number of live cells at day 0)/(number of cells at day 0) * 100%). The mean percentage of cell proliferation after 1 week of culture was as follows: SMCs: 95.5% (0 μg/mL SPIO), 94.3% (50 μg/mL SPIO), 93.6% (100 μg/mL SPIO), and 95.2% (200 μg/mL SPIO); ECs: 95.6% (0 μg/mL SPIO), 94.7% (50 μg/mL SPIO), 94.9% (100 μg/mL SPIO), and 97.1% (200 μg/mL SPIO).
The aforementioned results indicated reasonable to robust cell viability and proliferation in all groups after SPIO exposure. Thus, in an effort to maximize T2* MRI contrast, subsequent experiments concerning the SPIO-labeled SMC-EC cells (for histology, fluorescence microscopy, scaffold seeding, MRI) used an SPIO concentration of 200 μg/mL with a 48-hour incubation time. One exception to this occurred in the 2D cell culture, that is, the chamber slide, MRI experiments, where a shorter SPIO incubation time of 24 hours was also investigated to verify the effect of iron oxide exposure time on MRI contrast.
Intracellular Iron Oxide Uptake
After 1 week of in vitro cell culture, Prussian blue was found to stain positive (blue color) in intracellular regions of the cell cytoplasm for both SMC and EC lineages (Figure 2). In addition, positive red staining caused by the inherent fluorescent tag within the polymer microspheres indirectly confirmed intracellular iron oxide uptake (Figure 3). This is because the microspheres endocytosed by the cells also contained the SPIO microparticles.
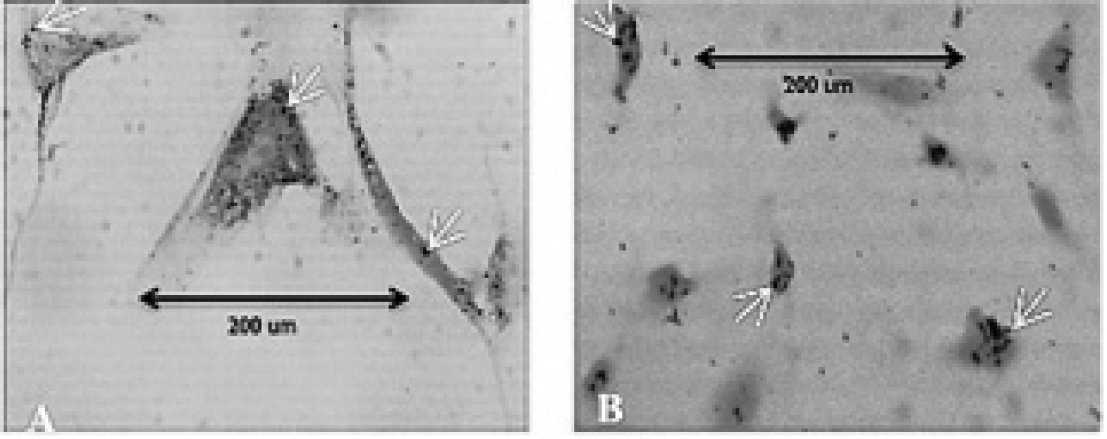
Prussian blue stain (X100 original magnification), with blue dots indicating the SPIO-rich regions. The cell cytoplasm was counterstained using nuclear fast red, as shown by the reddish-pink regions. (A) Smooth muscle cell and (B) endothelial cell populations, with arrows indicating evidence of intracellular iron oxide uptake.

Immunofluorescence of SPIO-labeled smooth muscle cells (A and B) and SPIO-labeled endothelial cells (C and D). Images were stained for F-actin (green) and DAPI (blue). The red fluorescence stain is derived from the microspheres (Bangs particles, Bangs Laboratories). As shown (E), both the iron oxide and the red fluorescent tag are centrally encapsulated in the core region of the polymer microsphere. Hence, red stains as seen in the figure provide indirect evidence of intracellular SPIO uptake as well.
Magnetic Resonance Imaging
We determined that vascular SMCs and ECs exposed to a concentration of 200 μg/mL of SPIO microparticles and 4.5 μg/mL of protamine sulfate with a 48-hour incubation time yielded maximum T2* contrast (Figure 4).
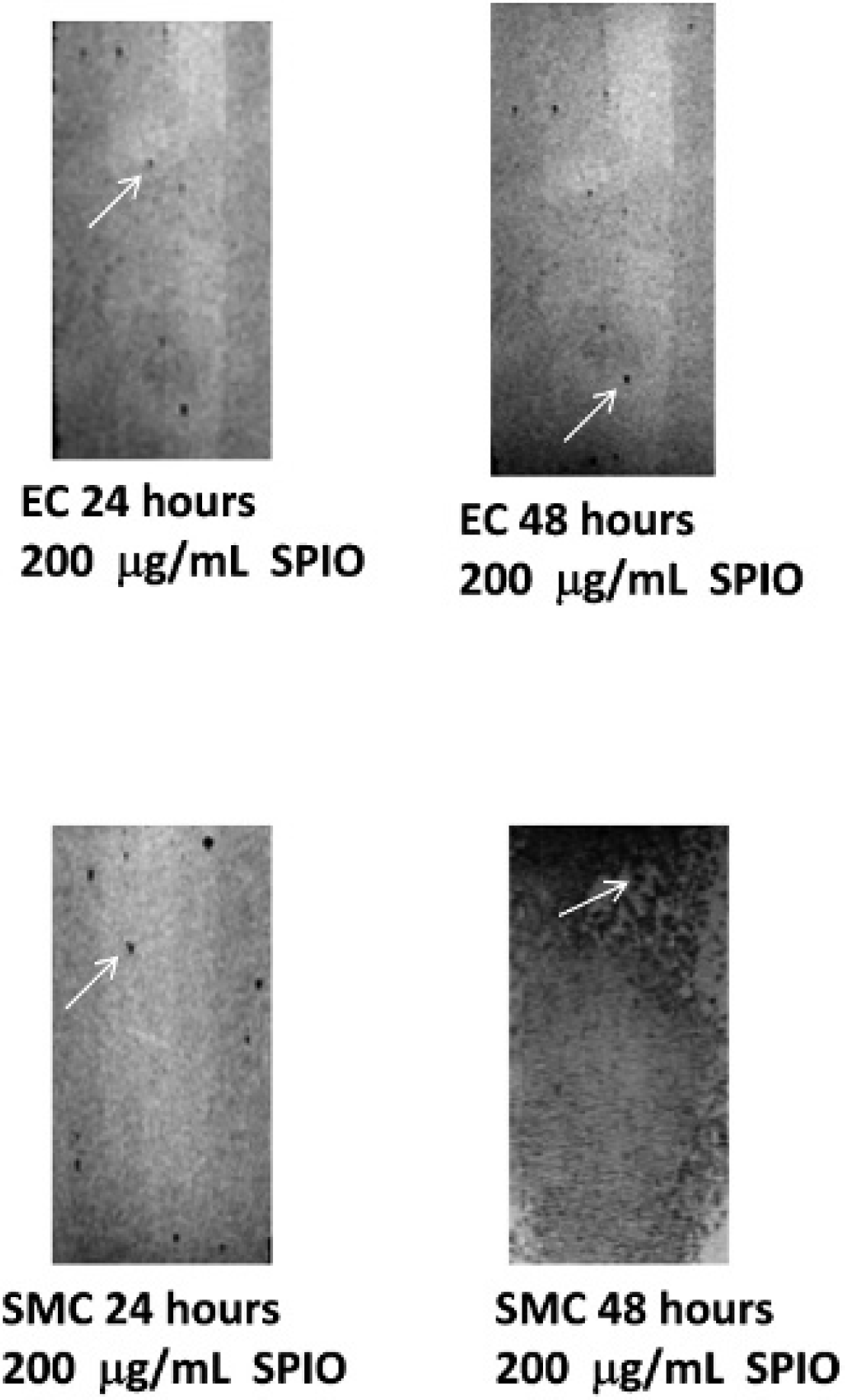
Hypointense T2* MRI contrast of SPIO-labeled endothelial cells (ECs) and smooth muscle cells (SMCs) at an SPIO concentration of 200 μg/mL and the use of 4.5 μg/mL of protamine sulfate transfection agent. ECs showed marginally improved contrast after 48 hours of incubation versus 24 hours. On the other hand, SPIO locations, which we believe to be mostly intracellular in origin (based on histologic evidence), appeared to be substantially darker in the case of SMCs when exposed to iron oxide for 48 hours. Arrows indicate hypointense regions of signal intensity, which we believe to be cells.
Intermittent cellular MRI was conducted over a 22-day period on cell-seeded, PGA/PLLA scaffolds (n = 3) that underwent steady flow conditioning at ≈ 5 to 6 dynes/cm2, which was physiologically relevant for heart valves. In the case of SMC-EC coculture–seeded scaffolds, no appreciable loss of T2* contrast was observed over the first 15 days (Figure 5A); however, there appeared to be considerable loss of hypointensity at 22 days. The MRI mean signal intensities (MSIs) at day 8, 15, and 22 regions of interest (ROI) were 57.2 (ROI area 0.24 cm2), 63.3 (ROI area 0.17 cm2), and 131.2 (ROI area 0.17 cm2), respectively (Figure 6A). However, other sections (Figure 5B) located a spatial location of 0.9 mm from the original images (see Figure 5A) appeared to have decreased in the size of hypointense regions, yet the MSIs measured temporally in these regions showed only marginal increases after 15 days (see Figure 5B): days 8, 15, and 22: 55.6 [ROI area 0.17 cm2], 81.5 [ROI area 0.14 cm2], and 89.6 [ROI area 0.05 cm2], respectively).

MRI showing the sagittal view of a representative smooth muscle cell–endothelial cell (SMC-EC)-seeded scaffold section exposed to steady flow (5–6 dynes/cm2). Specimens were secured by sample holders at each end. Hypointense regions of interest (ROI) are traced as shown. A, Loss in cellular SPIO contrast appeared to occur at 22 days of culture, that is, a temporal increase in mean signal intensity (MSI). This is likely due to the eventual response of the cells beyond 15 days of in vitro culture to the external steady flow mechanical stimulus, which was demonstrated by increased, out-of-plane cellular migration. The MSIs at day 8, 15, and 22 ROI were 57.2 (ROI area 0.24 cm2), 63.3 (ROI area 0.17 cm2), and 131.2 (ROI area 0.17 cm2), respectively. B, Another section spatially located 0.9 mm away from images in Figure 6A demonstrated a reduction in the areas of the ROI. However, the initial hypointensity observed at 8 days was sustained, even at 22 days. The MSIs at days 8, 15, and 22 were 55.6 (ROI area 0.17 cm2), 81.5 (ROI area 0.14 cm2), and 89.6 (ROI area 0.05 cm2), respectively.
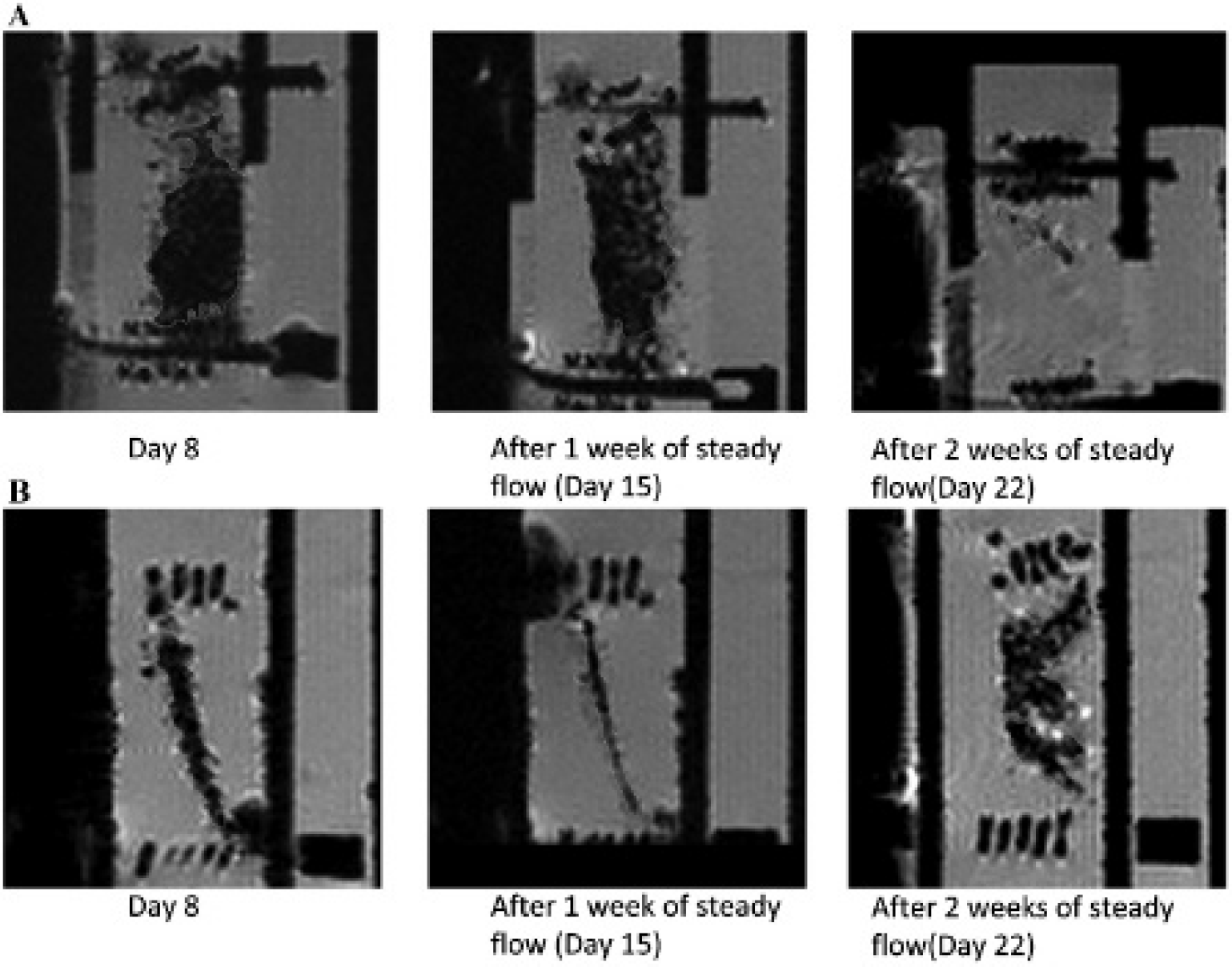
MRI showing the sagittal view of a representative bone marrow–derived mesenchymal stem cell (BMSC)-seeded scaffold section exposed to steady flow (5–6 dynes/cm2). Hypointense ROI are traced as shown. A, Consistent with the smooth muscle cell–endothelial cell (SMC-EC) specimens, loss in cellular SPIO contrast appeared to occur at 22 days of culture. However, distribution of hypointense signal intensity, which we interpret as more uniform distribution of the cells across the length of the scaffold, seemed to occur earlier at 15 days. The MSIs at day 8, 15, and 22 ROI were 54.7 (ROI area 0.79 cm2), 70.9 (ROI area 0.87 cm2), and 169.8 (ROI area 0.13 cm2), respectively. B, Yet, in another specimen, the size of the hypointense regions increased, with an eventual concurrent increase in SPIO contrast, that is, a temporal decrease in the mean signal intensity (MSI) at 22 days. The MSIs at day 8,15, and 22 ROIs were 120.8 (ROI area 0.35 cm2), 137.6 (ROI area 0.21 cm2), and 107.5 (ROI area 0.69 cm2), respectively. These observations reinforce the notion that longitudinal changes across any given cellular scaffold section subjected to steady flow occurred due to out-of-plane cellular migration rather than to accelerated SPIO expulsion from the cells.
A similar trend was observed for the BMSCs, wherein strong T2* contrast sustained itself over the first 15 days of tissue culture (see Figure 6A). The MSIs at days 8, 15, and 22 were 54.7 (ROI area 0.79 cm2), 70.9 (ROI area 0.87 cm2), and 169.8 (ROI area 0.13 cm2), respectively (see Figure 6A). However, at 15 days, the distribution of hypointense signal appeared spread out across the scaffold in the case of BMSCs compared to the SMC-EC-seeded scaffolds. Nonetheless, consistent with the SMC-EC specimens, substantial loss of hypointensity was observed with the BMSC-seeded scaffolds as well at 22 days. Yet in another BMSC-seeded specimen (Figure 6B), even though there was the appearance of two hypointense cell clusters at 22 days, which was also observed in the SMC-EC samples, the size of the sum of these hypointese regions increased (ie, a temporal decrease in MSI) at the end of 2 weeks compared to earlier time points. The MSIs at days 8, 15, and 22 were 120.8 (ROI area 0.35 cm2), 137.6 (ROI area 0.21 cm2), and 107.5 (ROI area 0.69 cm2), respectively (see Figure 6B).
DNA and Biochemical Content
The cellular DNA and biochemical content (collagen and GAGs) of the engineered tissues grown from unlabeled, SMC-EC co-culture–seeded, PGA/PLLA scaffold constructs were evaluated at the conclusion of the 22-day culture period.
DNA Quantification
The cellular DNA concentration was found to be 4,360 ng/g wet weight in the static group, whereas for the group exposed to steady flow, it was found to be 12,720 ng/g ± wet weight, that is, ≈ 2.92 times greater than that in the static group. The difference between the static and flow groups was found to be significant (p < .05) (Figure 7A).

(A) DNA content and (B) biochemical content of unlabeled smooth muscle cell–endothelial cell (SMC-EC)-seeded scaffolds after 3 weeks of incubation. The group exposed to flow exhibited a 99.5% increase in collagen compared to the static group (p < .05) (n = 3) (r = 5). Values presented in both A and B are the mean. Error bars indicate standard error of the mean. NS = nonnsignificant comparisons.
ECM Quantification for SMC-EC-Seeded Scaffolds
After a 3-week period, the collagen concentration was 436.8 ± 15.04 μg/g wet weight in the group exposed to steady flow compared to 218.9 ± 25.9 μg/g wet weight in the static group (Figure 7B). There was a 99.5% increase in collagen in the group exposed to flow compared to the static group (p < .05). On the other hand, the S-GAG concentration was 351.6 ± 44.5 μg/g wet weight in the group exposed to flow compared to 424.38 ± 36.6 μg/g wet weight in the control group. This represents a 17.1% decrease in GAG content in the scaffolds exposed to steady flow in comparison with the static controls (see Figure 7B) but was found not to be significantly different (p > .05).
Discussion
TEHVs appear to be a promising technology in the treatment of severe congenital valve anomalies and are appealing in that the constructs may potentially grow and continuously remodel with the patient. Several studies have already demonstrated that remodeling events recapitulate distinct valvular layers and can enable in vivo functionality.7,25–34 Shinoka and colleagues showed that autografts are preferable over allografts. 33 Sutherland and colleagues were able to demonstrate contiguous normal valve functionality (negligible regurgitation) and extensive remodeling in sheep using a tissue-engineered heart valve approach using BMSCs. 7 Meanwhile, O'Brien and colleagues presented decellularized tissue scaffolds as an alternative approach that permitted recellularization by animal host fibroblastoid cells. 29 A recent review by Schoen delineated the ability of TEHVs to permit somatic growth, thereby making the overall TEHV strategy appealing for the treatment of severe congenital valve disease.32 Indeed, although clinical translation has yet to occur, in vivo evidence that supports the proof of concept in this approach is overwhelming. However, TEHV design specifics for scaffold properties, choice of cell source, and construct optimization through in vitro mechanical conditioning need further fine-tuning before an accepted protocol can be applied clinically. Additionally, a key to this translation lies in effective noninvasive and nondestructive monitoring of the fate of implanted cells. Moreover, tissue remodeling during initial phases after implantation will depend on cell migratory events to and from the scaffold and will involve both the cells used to seed the scaffolds and the endogenous cells of the artery. Relevant native cell sources are human artery SMCs and ECs, whereas BMSCs have been extensively explored as a single autologous cell source for heart valve tissue engineering.5–7,35
In line with the importance of monitoring cellular activity in TEHVs, we were motivated in understanding the effect of mechanical environments on cell migratory events within the scaffold, which, to our knowledge, has not been systematically investigated to date. In this context, the hemodynamic environments that valves are subjected to, such as fluid-induced shear stresses, can affect migratory events. Our previous work examined static environments and found the migratory events of vascular cells to be negligible. 21 As an initial approach, here we observed that the introduction of a steady flow component offers remarkable alterations in cell migratory behavior compared to static environments over a similar time frame. This was seen using SPIO-based cellular MRI, both in terms of temporal changes in intrascaffold vascular cell distribution and in the loss of hypointense signal intensity with time. We interpret the latter as out-of-plane cell migration rather than loss of contrast due to cell proliferation because regions of robust, hypointense T2* MRI contrast within the scaffold environment in static culture were still observed for up to 12 days in our previous work. 21 Nonetheless, here we exposed the SMCs and ECs to > 15 times higher SPIO concentrations compared to the amount previously used 21 and found that the cells were still reasonably viable (at 200 μg/mL SPIO concentration: SMCs 74.8% and ECs 87.9%) and could proliferate well (at 200 μg/mL SPIO concentration: SMCs 95.2% and ECs 97.1%). Finally, reduced hypointensity in the samples exposed to steady flow occurred only after the first 15 days, whereas the rate of cell proliferation would generally be higher during the initial, rather than the latter, culturing period. It is also important to point out that even though fluid-induced shear stresses could presumably invoke expulsion of iron oxide from cells, we do not believe this to be the case for the following reasons. Sample sections (see Figure 5B) located a distance of 0.9 mm away from an initial image set (see Figure 5A) showed that the signal intensity of hypointense regions does not decrease over time even though the size of the hypointense areas decreases. This suggests that two effects can occur temporally under shear stress environments, namely, that cellular contrast can decrease and that the size of hypointense regions can also reduce. Although the former may point to the potential expulsion of iron oxide, the latter cannot because the signal intensities did not change considerably over time. Indeed, the MSIs at days 8, 15, and 22 did not change substantially after 15 days (see Figure 5B: MSIs at days 8, 15, and 22: 55.6 [ROI area 0.17 cm2], 81.5 [ROI area 0.14 cm2], and 89.6 [ROI area 0.05 cm2], respectively). In addition, although BMSC-seeded specimens yielded some similar trends to SMC-EC samples (loss of contrast [see Figure 6A] and two cell clusters at 22 days [see Figure 6B]), the size of the hypointense regions was much larger than the preceding time points without having lost T2* contrast, which would have been possible only if cells migrated into the section (see Figure 6B: MSIs at days 8, 15, and 22 were 120.8 [ROI area 0.35 cm2], 137.6 [ROI area 0.21 cm2], and 107.5 [ROI area 0.69 cm2], respectively). Furthermore, two previous studies of SPIO-labeled cells exposed to fluid-induced shear stress demonstrated robust T2* or T2 contrast at the conclusion of time scales comparable to our study.36,37 These observations in sum led us to believe that it is more likely that the alterations to the hypointense regions under steady flow were due to out-of-plane cellular migration rather than accelerated iron oxide expulsion or proliferation-related intracellular SPIO dilution, which may have resulted from provocation by the local shear stress environment.
We also found that SPIO-labeled BMSCs also behaved in a similar manner under steady flow, with cell colonies appearing to spread across and migrate out of plane (see Figure 6). Therefore, flow-based environments seem to promote the migratory activity of cells in scaffolds intended for the heart valve tissue engineering application by spreading seeded cells across and through the thickness of the scaffold. The increased cell migration occurred as a direct result of the fluid-induced mechanical cues received, which was previously not evidenced in cellular constructs cultured in static conditions. 21 That said, our study has several limitations. First, the cell signaling pathway and/or activity have yet to be delineated. Specifically with regard to activity, we did not assess the relative flow-induced migration patterns of the ECs versus the SMCs within the scaffolds, nor did we explore the differentiation states of the BMSCs; these are areas that are currently being investigated in our laboratory. Second, we did not establish statistical significance between SPIO-labeled and unlabeled cellular counterparts; rather, we only observed that SMCs and ECs were able to remain reasonably viable and proliferate well even after being subjected to high concentrations of iron oxide (200 μg/mL). As such, further studies are needed to optimize the SPIO microparticle concentrations being used to label these cell types (vascular ECs, vascular SMCs, and BMSCs). Third, although we observed that the cellular PGA/PLLA scaffolds that were analyzed after steady flow conditioning did show retention of intracellular iron oxide content (positive Prussian blue histologic staining; data not shown), the evidence was preliminary, and more experiments are needed to confirm this finding. Despite these limitations, the results found here on steady flow–mediated intrascaffold migratory events, as observed by longitudinal cellular MRI, may explain the reason for enhanced cellularity and ECM quality that is traditionally associated with engineered heart valve tissues exposed to fluid flow in comparison with static culture groups,5,23 which we also found to be the case in this study (see Figure 7). Last but not least, our intention here was to ensure that physiologic shear stresses were imparted onto the growing specimens, which we have previously established occurs at the shear stresses that were applied, that is, 5 to 6 dynes/cm2.12,23,24 However, the actual flow physics on the samples can be effectuated only with the use of a pulsatile arterial flow waveform, which may cause altered cellular distributions compared to the steady flow state that was investigated here and is thus another limitation of our study. The novelty of this work is that we are, to our knowledge, the first to report on the noninvasive MRI tracking of SPIO-labeled cells within evolving tissue constructs exposed to a physiologically relevant (shear stress) mechanical conditioning (steady flow) environment, which is especially important for cardiovascular and heart valve tissue engineering studies. Thus, despite the aforementioned limitations, the protocols established here will be useful for in vitro research and development protocols in the area of heart valve tissue engineering in assessing intrascaffold cell migration and redistribution over time.
Summary
We conclude that vascular cells (SMCs and ECs) can be labeled with SPIO microparticles in a reasonably efficient manner and at much higher concentrations compared to what has previously been shown, 21 thereby providing stronger, sustained MRI contrast. Moreover, steady flow mechanical conditioning enhances cell migration processes and the uniformity in cell distribution across and through the thickness of the scaffold, which are likely to have been the reasons for the increased cellularity and biochemical content (eg, collagen) findings typically found in studies involving flow versus no flow conditions.5,23,38 Finally, the SPIO-based MRI methods reported in our study can be used in heart valve tissue engineering efforts to understand how cell distributions are altered under physiologically relevant stresses for heart valves, such as from flow, flexure, and stretch sources, or any combination of these mechanical stimuli.
Footnotes
Acknowledgments
We thank Dr. Daniel Pelaez at the University of Miami for his assistance with the use of an inverted florescent microscope.
Financial disclosure of authors: Funding for this work was provided by American Heart Association Scientist Development Grant 0830061N.
Financial disclosure of reviewers: None reported.