
Abstract
We present a flexible and highly reproducible method using three-dimensional (3D) multicellular tumor spheroids to quantify chemotherapeutic and nanoparticle penetration properties in vitro. We generated HeLa cell–derived spheroids using the liquid overlay method. To properly characterize HeLa spheroids, scanning electron microscopy, transmission electron microscopy, and multiphoton microscopy were used to obtain high-resolution 3D images of HeLa spheroids. Next, pairing high-resolution optical characterization techniques with flow cytometry, we quantitatively compared the penetration of doxorubicin, quantum dots, and synthetic micelles into 3D HeLa spheroid versus HeLa cells grown in a traditional two-dimensional culturing system. Our data revealed that 3D cultured HeLa cells acquired several clinically relevant morphologic and cellular characteristics (such as resistance to chemotherapeutics) often found in human solid tumors. These characteristic, however, could not be captured using conventional two-dimensional cell culture techniques. This study demonstrated the remarkable versatility of HeLa spheroid 3D imaging. In addition, our results revealed the capability of HeLa spheroids to function as a screening tool for nanoparticles or synthetic micelles that, due to their inherent size, charge, and hydrophobicity, can penetrate into solid tumors and act as delivery vehicles for chemotherapeutics. The development of this image-based, reproducible, and quantifiable in vitro HeLa spheroid screening tool will greatly aid future exploration of chemotherapeutics and nanoparticle delivery into solid tumors.
IN RECENT YEARS, three dimensional (3D) culture systems have gained increasing recognition as an effective tool for biologic research. 3D cultured cells more closely mimic the physiologic environment of living organisms compared to conventional monolayer culture systems. 1 Multicellular tumor spheroids are one of the classic 3D culture models that can be applied to the development of anticancer drugs and treatments. By mimicking the 3D network of the cellular–matrix and cell-cell interactions, tumor spheroids resemble many aspects of the pathophysiologic environment within human tumor tissue. Multicellular tumor spheroids also mimic in vivo tumor-like development patterns of avascular tumor nodules in terms of morphology and growth kinetic properties.2–4 Additionally, xenograft tumor sections from nude mice and tumor spheroids appear to have similar hematoxylin and eosin staining. Therefore, a thorough understanding of tumor spheroids is indispensable for biologic research, especially for the prospect of cancer therapy development.
Nevertheless, a large number of cell culture studies are still being conducted using monolayer cell cultures. Monolayer cell cultures are easily maintained and monitored. No living multicellular organisms, however, exist solely in a two-dimensional (2D) space. Therefore, 3D cultures have a closer resemblance to cells growing in the in vivo tissue environment. The replacement of 2D cell culture with 3D tumor spheroids is not an easy task. Growing tumor spheroids under artificial conditions is not challenging, and several mature procedures exist2,3,5; the difficulties reside in how to thoroughly characterize 3D tumor spheroids. Standardized characterization methods to categorize spheroid morphology are still lacking due to difficulties in imaging 3D structures.
Our interest in developing a new anticancer nanomedicine made evident the lack of reliable model methods to visualize and analyze the efficacy of novel anticancer therapeutics. Therefore, we developed a series of imaging-based characterization techniques to examine HeLa cell–derived 3D tumor spheroids. Our goal was to establish clear, qualitative, and quantitative characterization methods of tumor spheroid morphology. The imaging techniques used to visualize the external and internal structures of 3D cultures in this study included scanning electron microscopy (SEM) and transmission electron microscopy (TEM), respectively. For stereo visualization of HeLa spheroids, multiphoton microscopy followed by 3D reconstruction of the image sections was performed.
With these high-resolution imaging methods, we established precise surface topography of the HeLa cellular spheroids and revealed characteristic internal structures of these spheroids. The HeLa 3D spheroid closely resembled the in vivo avascular tumor nodular appearance, suggesting that 3D spheroid cell culture systems can functionally mimic tumor nodular properties. To visualize the penetration behavior of anticancer therapeutics, such as doxorubicin, quantum dots, and micelles, detailed multiphoton microscopy data were collected and used to reconstruct 3D images of HeLa spheroids. Our study results demonstrate that by using 3D imaging approaches, we are able to gain insights into chemotherapeutic penetration capabilities and set a precedence for HeLa spheroids as an efficient initial screening method to aid future anticancer therapy development.
Materials and Methods
Construction of Multicellular Tumor Spheroids
The HeLa (human cervical adenocarcinoma) cellular spheroid model was constructed using the liquid overlay method. Briefly, HeLa monolayer cells were cultivated in Dulbecco's Modified Eagle's Medium (DMEM low glucose) (Hyclone, Beijing, China) containing 10% (vol:vol) fetal bovine serum (FBS) (Hyclone) supplemented with antibiotic/antimycotic mix (Hyclone). When the cells reached about 70% confluence, they were harvested from the culture dishes by 0.25% (wt:vol) trypsin–0.53 mM ethylenediaminetetraacetic acid (EDTA) (Gibco, Grand Island, NY) and then suspended in DMEM supplemented with 15% FBS. Single-cell suspension was seeded to flat-bottomed 96-well plates previously coated with 1% (wt:vol) agarose that prevented cell adhesion to the wells. The inoculation concentrations of the HeLa cells were 62, 125, 250, 500, 1,000, and 2,000 per well. After that, the HeLa cells were incubated in a humidified atmosphere with 5% CO2 in air at 37°C for about 10 days, and the culture medium was changed every other day.
Characterization of HeLa Cellular Spheroids by Optical Microscopy
Spheroid integrity can easily be visualized by phase-contrast imaging, which is also the basis for recording spheroid volume growth kinetics. Images for quantitative analysis of the size distribution and shape of HeLa cellular spheroids were captured at the desired time points using a Leica (Hong Kong, China) DMI3000 microscope fitted with a cooled charge-coupled device camera. In the case of HeLa cellular spheroids, the projection area, A, and the perimeter of the projected area, P 5 , were determined for each spheroid using a Leica IMAGE version workstation. The data were used to calculate shape factor and corrected volume, which define the sphericity and real volume of HeLa cellular spheroids, respectively. To measure the growth of the HeLa cellular spheroids, we collected 10 to 20 spheroids for each sample, which were cultured for various time periods and detached, and counted the cell number using a cell counting machine (Vi-Cell XR, Beckman Coulter, US).
Histologic Analysis of HeLa Cellular Spheroids by Hematoxylin and Eosin Staining
Intact HeLa cellular spheroids were collected at desired time points and fixed by 10% formaldehyde solution for 24 to 48 hours. For the embedding and cutting steps, HeLa cellular spheroids were dehydrated by an ethanol gradient from 50% up to 100%. The spheroids were further dehydrated with 100% ethanol followed by verification with xylene (30 minutes × 2) and impregnation with paraffin wax (60 minutes × 2). Then the samples were embedded into paraffin blocks and cut by a Leica Ultracut UCT microtome (Leica EM UC6) into 8 μm paraffin ribbons. For the staining process, the sections were stained with alum hematoxylin and rinsed by running water. After being differentiated by 0.3% acid alcohol and rinsed, the sections were stained with eosin for 2 minutes. Finally, the sections went through a series of dehydration, clearing, and mounting and were then observed by optical microscopy (DMI3000, Leica).
Live/Dead Assay of HeLa Cellular Spheroids by Flow Cytometry
We present a quantitative method for optimal observation of the internal structure of HeLa cellular spheroids using the LIVE/DEAD Viability/Cytotoxicity Kit (calcein AM and ethidium homodimer [EthD-1], Invitrogen, Grand Island, NY) by flow cytometry. Briefly, for the quantitative method by flow cytometry, appropriate numbers of HeLa cellular spheroids were collected and digested by trypsin-EDTA to single-cell suspension, which were washed by phosphate-buffered saline (PBS) to remove or dilute serum esterase activity. The cells were stained by calcein AM (0.5 μM) and EthD-1 (8 μM) for 20 minutes at room temperature prior to flow cytometry measurement (BD Biosciences, US). Samples were excited at 488 nm, and green fluorescence emission for calcein (ie, 530/30 bandpass) and red fluorescence emission for EthD-1 (ie, 610/20 bandpass) were recorded.
Cell-Cycle Analysis of HeLa Cellular Spheroids and HeLa Monolayer Cells by Flow Cytometry
HeLa monolayer cells were seeded in 100 mm culture dishes and incubated for 48 hours, whereas HeLa cellular spheroids were generated as described previously. Cells of monolayer and spheroids were harvested at desired time points, dissociated, fixed in precooled (4°C) ethanol (70%), and stored at −20°C for at least 2 hours. After removing ethanol, the fixed cells were treated with a PBS staining solution containing propidium iodide (100 μg/mL) and ribonuclease (50 μg/mL) for about 20 minutes. Stained cells were analyzed with the BD FACS Calibur Flow Cytometry System (BD Biosciences).
Superficial Morphology of HeLa Cellular Spheroids by SEM
Intact HeLa cellular spheroids at different time points were selected from flat-bottomed 96-well plates, whereas monolayer cells were detached and collected. Then the sampled cells were washed by PBS (pH 7.4), fixed overnight at 4°C in 5% glutaraldehyde with 0.15 M PBS, and then washed with 0.15 M PBS three times. After that, spheroids were progressively dehydrated by a gradient concentration of ethanol series, isoamyl acetate for 0.5 hours, followed by critical point drying with carbon dioxide. Finally, the samples were observed by field emission SEM without gold coating to display the actual morphology of the spheroids.
Internal Structure of HeLa Cellular Spheroids by TEM
HeLa cellular spheroids (perfectly) and monolayer cells (dissociated) were collected for sample preparation. Primary fixation was done with 3% glutaraldehyde followed by secondary fixation with 1% osmium tetroxide in dark for 2 hours at room temperature. The flat layers and spheroids were suspended in 2% molten agar to make small blocks. The solidified agar blocks were dehydrated by ethanol gradient. For the embedding step, the dehydrated agar blocks were suspended in propylene oxide followed by treatment with 1:1 mixture of propylene oxide and epoxy resin (Cy-812). After a succession of processing, the blocks were placed in a refined beam capsule for polymerization. Next, the resin blocks were carefully trimmed. Sections of consistent thickness (90 nm) were cut using a Leica Ultracut UCT microtome (EM UC6) followed by uranyl acetate staining. Sections on the grids were observed using an electron microscope (JEOL, JEM-1400) at 80 kV.
Tomographic Scanning of Living HeLa Cellular Spheroids by Multiphoton Fluorescent Microscopy
We established a qualitative method to observe the internal structure of living HeLa cellular spheroids with 4′6-diamidino-2-phenylindole (DAPI) (Invitrogen) by multiphoton fluorescent microscopy. Briefly, HeLa cellular spheroids were prestained by DAPI (50 mg/mL) after incubation overnight at 37°C. Before observation by multiphoton fluorescent microscopy (Leica TCS MP5 multiphoton (MP) fluorescent correlation spectroscopy (FCS) microscope), the spheroids were washed with PBS to remove redundant dyes, which can increase the signal to noise ratio effectively. To obtain the greatest sensitivity using laser scanning microscopy, a fluorescent optical filter (405 ± 10 nm) was chosen for DAPI.
Doxorubicin Penetration Analysis of HeLa Monolayer Cells and Cellular Spheroids
To observe the process of doxorubicin entering the 2D cultured cells, laser confocal fluorescent microscopy was used. Briefly, HeLa monolayer cells were seeded in confocal dishes (Corning) and cultured for about 12 hours. After adding doxorubicin (10 μg/mL), fluorescent images were obtained at 5 minutes, 30 minutes, and 3 hours. After combining the fluorescent images with corresponding bright field images and colocalizing the images, the entering process of doxorubicin could easily be tracked.
We used multiphoton fluorescent microscopy and flow cytometry to analyze the penetration of doxorubicin into HeLa cellular spheroids qualitatively and quantitatively. After being exposed to doxorubicin (10 μg/mL) for 5 minutes, 30 minutes, 1 hour, 2 hours, 4 hours, and 12 hours, HeLa cellular spheroids on the fifth day were photographed by multiphoton fluorescent microscopy (TCS SP5, MP FCS) to trace the penetration of doxorubicin. At the same time, the HeLa cellular spheroids were collected, digested to cell suspension, and then analyzed directly by flow cytometry (Beckman Coulter) to quantify the amount of doxorubicin taken up by the spheroids.
Quantum Dot Penetration Analysis of 2D and 3D Cultured HeLa Cells
To investigate the delivery of two types of quantum dots (amino PEG Qdot605 and carboxyl Qdot605, Invitrogen), the HeLa cells were cultured as above in confocal dishes and agarose-coated 96-well plates for 2D and 3D culturing, respectively. After 24-hour treatment with quantum dots (20 nM, diluted with serum-free medium), the flat layers and spheroids were examined by fluorescence microscopy (Leica, DMI 3000) and multiphoton fluorescent microscopy (TCS SP5, MP FCS).
Doxorubicin-Loaded Micelle Penetration Analysis of HeLa Monolayer Cells and Spheroids
We established qualitative and quantitative methods to study the penetration processes of three types of doxorubicin-loaded micelles. Briefly, the cells were seeded for 2D and 3D culture and treated with 10 mg/mL doxorubicin encapsulated in micelles suspended in culture medium for 4 hours. After that, the samples were examined by laser confocal fluorescent microscopy (Nikon) and multiphoton fluorescent microscopy (TCS SP5, MP FCS) for qualitative characterization or dissociated and determined by flow cytometry (Beckman Coulter).
Results
Morphologic Characterization of HeLa Spheroids
We generated HeLa spheroids using the liquid overlay method.2,4 Even when seeded at different densities, the spheroids had similar growth trends measured by spheroid diameter and corrected volume 5 (Figure 1A). Spheroids were imaged at regular time intervals, and their growth kinetics and collapsed end points were recorded (see Figure 1A). We also recorded spheroids versus monolayer cell counts from the same seeding densities (Figure 1B). From the growth curves of 2D versus 3D cultured HeLa cells, it was apparent that the doubling time of spheroids was longer than that of monolayer cells. The doubling time of HeLa spheroids corresponded to the doubling time of in vivo solid tumors, demonstrating that HeLa multicellular spheroids mimicked the growth kinetic properties of in vivo tumors.6,7

A, Optical characterization of HeLa cellular spheroids: (1) diameters, (2) shape factors, (3) corrected volumes, and (4) optical imaging (scale bars 200 μm) of HeLa spheroids with seeding densities of 62 cells/well, 125 cells/well, 250 cells/well, 500 cells/well, and 1,000 cells/well at different time points. B, The growth curves of HeLa cellular spheroids (1) and monolayer cells (2). C, The external morphology of intact HeLa cellular spheroids (250 cells/well) and adhesive monolayer cells at different time points observed by scanning electron microscopy without gold coating.
To evaluate the surface morphology of HeLa cellular spheroids versus HeLa monolayer cells, field emission SEM was performed (Figure 1C). In the intact spheroid, every cell formed a spherical morphology, and filopodia were visible surrounding the cells. The 2D cultures conversely remained as flat layers, fused together via supporting connective structures. These morphologic characteristics of HeLa tumor spheroids further demonstrated their morphologic similarities to in vivo solid tumors.5,8 Next, we aimed to focus on the internal proliferative status of HeLa spheroids.
Cell Proliferative Characteristics
To better characterize the internal structures of HeLa spheroids, wax sections were stained by hematoxylin and eosin (to visualize cellular nuclei and cytoplasm). Based on the staining patterns, individual cells from the HeLa spheroids can be divided into three distinct populations: proliferative rim, quiescent region, and necrotic core (Figure 2A). For quantitative analysis, cell viability was measured by flow cytometry after staining with calcein AM and EthD-1 to discriminate cellular viability status. From flow analysis, the cells of spheroids were also clearly distinguished into three populations (proliferative rim, quiescent region, and necrotic core), similar to the findings using hematoxylin-eosin staining (see Figure 2A). These results provide both qualitative and quantitative evidence of HeLa spheroids mimicking in vivo solid tumor properties in addition to providing evidence of the induction of cell necrosis within the multicellular areas far from nutrient and oxygen supplies, such as the center core of the spheroids.9–11
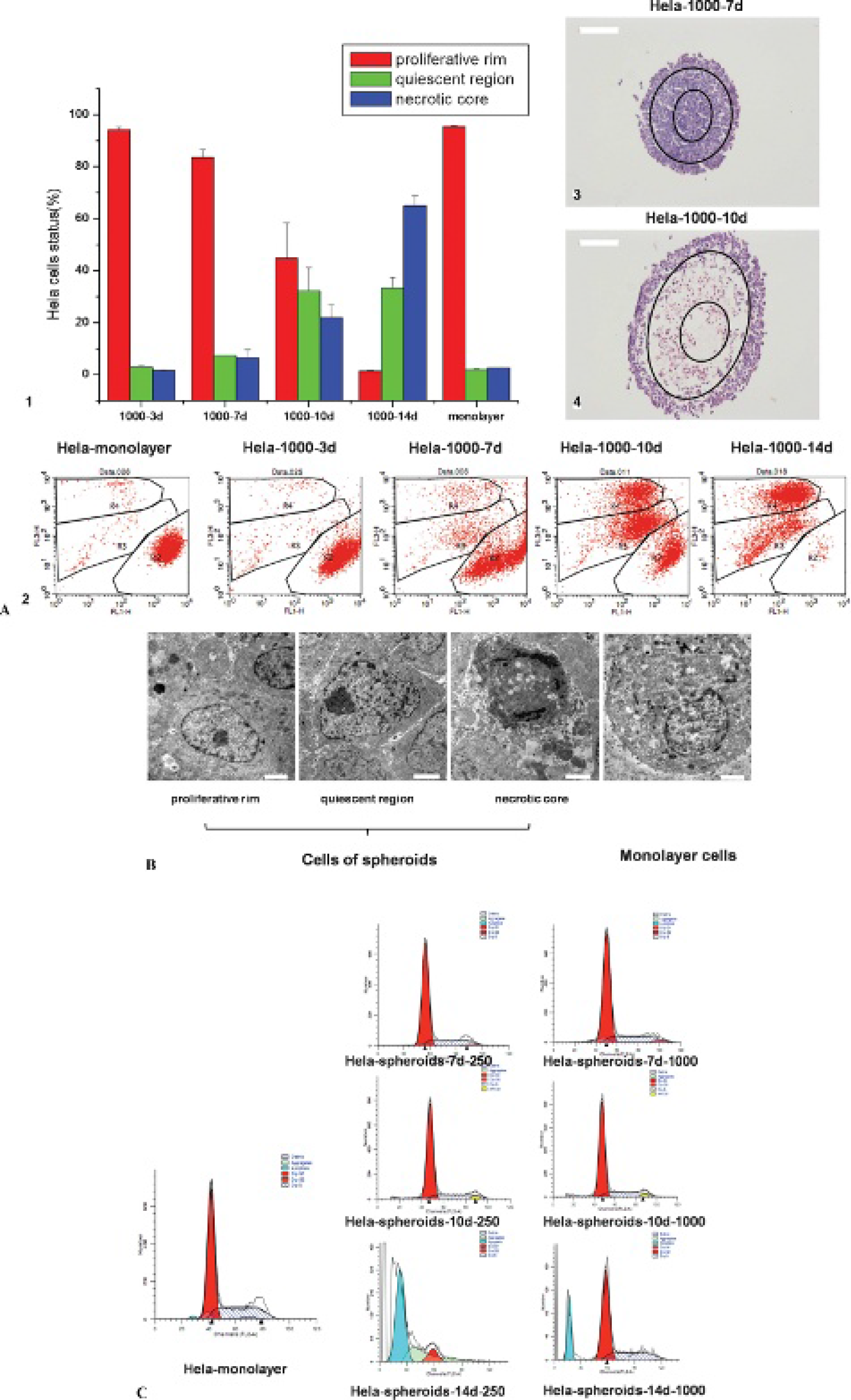
A, Cell status analysis by flow cytometry and hematoxylin-eosin (H&E) staining. (1) and (2) Live/dead assay. The dots in regions R2, R3, and R4 represent the cells of the proliferative rim, which were all stained by calcein; the cells of the quiescent region, which were stained by calcein and EthD-1; and the cells of the necrotic core, which were stained by EthD-1 very deeply, respectively. (3) and (4) H&E staining of HeLa spheroids with 250 cells/well and 1,000 cells/well primary inoculation. B, Internal morphology of HeLa cellular spheroids and monolayer cells observed by transmission electron microscopy (scale bar 2 μm). C, Cell-cycle analysis of HeLa cellular spheroids and monolayer cells.
To further characterize the spheroid core, we performed TEM of both HeLa spheroids and HeLa monolayer cells (Figure 2B). The images supported hematoxylin-eosin staining and cell survival assay data of spheroid cells separating into three cell types of the proliferative rim, quiescent region, and necrotic core. In the compact spheroids, we found cells in asynchronous proliferative, quiescent, and necrotic states. Cells in the proliferative state contained intact nuclei and abundant microvilli, which suggested optimal intra- and extracellular conditions. Cells in the quiescent stages, however, contained shrunk nuclei and sunken-in cell membrane morphology. Cells undergoing necrosis contained disintegrated nuclei and cellular membrane morphology. In the 2D cultures, however, all the HeLa cells were in the proliferative state and had intact nuclei and cell membranes (see Figure 2B).
Considering the difference in proliferation status of 2D and 3D cultured cells, cell-cycle analysis was conducted to determine the deoxyribonucleic acid (DNA) content of monolayer versus spheroid cells. It was striking how fewer cells in 3D cultures were in S phase compared to 2D cultured cells (Figure 2C); this suggests that 3D cultured HeLa cells have growth kinetics similar to that of vivo tumors.6,7 These data further provide evidence that the center of spheroids is mainly populated by quiescent cells (see Figure 2A). Interestingly, after 14 days in culture, spheroids became destabilized due to cellular apoptosis (see Figure 2C), which also confirmed data obtained from growth curves showing a reduction in cell count at the end of the incubation period.
3D Observation of HeLa Cellular Spheroids
There are significant technical challenges, however, to the microscopic study of living tumor spheroids at increasing imaging depths due to increased light scattering. 12 To address this issue, we established a unique method for 3D imaging of living HeLa cellular spheroids by multiphoton microscopy. After staining, individual spheroids from HeLa 3D cultured samples were photographed every 5 mm section from the top to the bottom. The 3D images of the spheroids were reconstructed using tomography (Figure 3). The whole structures of living and in vivo-like tumor spheroids were directly achieved by these 3D images and demonstrated the potential to use this 3D imaging technique to gain insight into the penetration properties of chemotherapeutics and nanoscale image materials used in clinical settings.

3D images of HeLa spheroids. The images of tomographic scanning (left) were taken every 5 μm section from the top to the bottom of an intact spheroid, whereas the 3D image (right) was reconstructed using tomography.
Doxorubicin Penetration Analysis
We applied our 3D imaging method of tumor spheroids to study the penetration behavior of doxorubicin, one of the most celebrated chemotherapeutics, commonly used to treat a wide range of cancers. When HeLa cells were grown in a monolayer, doxorubicin was absorbed rapidly during a short period of time. The red region stained by doxorubicin extended from the cell membrane into the cytoplasm and eventually reached the nuclei (Figure 4A). We compared the penetration of doxorubicin into monolayer cells versus spheroids and found that doxorubicin entered spheroids much more slowly than an equivalent number of monolayer cells (Figure 4B). This demonstrated that HeLa cells in 3D structures provide a multicellular resistance model that mimics the chemotherapy resistance often found in solid tumors in vivo.13–16

Penetration of doxorubicin into (A) HeLa monolayer cells and (B) HeLa cellular spheroids.
Penetration Analysis of Quantum Dots and Micelles
In addition to doxorubicin, we applied our 3D imaging method to visualize the delivery process of water-soluble quantum dots and doxorubicin-encapsulated micelles. We used two types of quantum dots, amino PEG Qdot605 and carboxyl Qdot605 (Invitrogen), which had the same size and hydrophobicity but different charges, to treat HeLa monolayer cells and spheroids. In monolayer cells, the two quantum dots had slightly different, but otherwise very similar, intracellular fluorescence intensity. The cells treated with the amino PEG Qdot605 exhibited a slightly stronger fluorescence than the carboxyl PEG Qdot605–treated cells (Figure 5). In HeLa cellular spheroids, the penetration of these two quantum dots was dramatically different: the positively charged quantum dots barely penetrated into spheroids, whereas the negatively charged nanoparticles were able to travel deeply into the cells of spheroids (see Figure 5 and Figure S1 available in the online version only).

Penetration of quantum dots into (A) HeLa monolayer cells and (B) HeLa cellular spheroids.

The measurement of penetration of negatively charged quantum dots into HeLa cellular spheroids (scale bar 200 μm). The distance from the center of the spheroid to the boundary of the penetrating quantum dot is indicated as r, and the distance from the center to the periphery of the spheroid is indicated as R.
Next, we preformed qualitative and quantitative analysis to study the penetration of three types of doxorubicin-loaded micelles: methoxy polyethylene glycol-poly(lactic-co-glycolic)acid (mPEG-PLGA), gelatin-polylactic acid (Gel-PLA), and poly(L-aspartic acid-co-L-lactide)-1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (PAL-DPPE) in the 2D and 3D cultured HeLa cells. These micelles were constructed from different polymer building blocks. In the monolayer cells, the intracellular fluorescence intensity of mPEG-PLGA- and PAL-DPPE-treated cells were almost the same, whereas the Gel-PLA-treated cells had weaker intracellular fluorescence (Figure 6). In the 3D culture system, mPEG-PLGA-treated cells had moderate fluorescence, whereas PAL-DPPE-treated cells had the strongest fluorescence, which indicated deeper penetration, and Gel-PLA-treated cells had the weakest fluorescence, presenting the shortest translocation (see Figure 6).

Penetration of micelles into (A) HeLa monolayer cells and (B) HeLa cellular spheroids.
Discussion
In this study, we established an imaging-based method to study the delivery of chemotherapeutics and nanoparticles in a 3D tumor model—multicellular HeLa spheroids. By using 3D multiphoton microscopy paired with conventional flow cytometry and histologic techniques, we were able to quantitatively characterize the morphology of HeLa spheroids. Our results indicate that the HeLa spheroid model system closely resembled in vivo avascular tumor nodular appearance and contained a cellular center (necrotic core). The necrotic core was degenerate, whereas the culture medium contacting outer layer cells (proliferative rim) resembled the highly proliferative in vivo solid tumor cells located near nutrient-rich capillaries.9–11
Furthermore, we established SEM and TEM 3D image reconstruction techniques to characterize the external and internal structures of 3D versus 2D culture systems. These techniques made it possible for detailed morphologic distinctions between the two culture systems. This approach directly revealed in-depth topography of the 3D cultures at nanoscale resolution, which clearly resembled in vivo tumor-like morphology.17–19 Two-photon or multiphoton microscopy is fundamentally different from traditional linear excitation microscopy. Multiphoton microscopy uses higher-order light–matter interactions involving multiple photons for contrast generation.20–22 By using multiphoton microscopy, we were able to reconstruct 3D images of the topography of living spheroids. It is well understood that imaging spheroids by optical microscopy is technically difficult due to high levels of light scattering in deep 3D cellular structures. 20 Therefore, multiphoton microscopy greatly improves image resolution. At this higher image resolution, the biologic effects of chemotherapeutics or nanomaterials on 3D tumor spheroids, previously unseen by conventional light microscopy, can now be visualized.
With the multiphoton microscopy techniques, we tested the penetrability of nanoparticles (quantum dots) and micelle-encapsulated doxorubicin into HeLa spheroids. The intracellular localization of quantum dots depends on a variety of factors: surface charge, size, and functionality.23–25 Previous studies demonstrated that quantum dots have great potential as effective delivery vehicles for chemotherapeutics or imaging materials.23,26–28 It is generally believed that positive surface charges may enhance quantum dot internalization.29,30 Our results demonstrated that negatively and positively charged quantum dots had slightly different, but otherwise very similar, uptake behavior in HeLa cells grown in flat monolayers, although the positively charged quantum dots did exhibit a slightly stronger intracellular fluorescence than the negative charged particles (see Figure 5). In the 3D cultures, however, both positively and negatively charged quantum dots had reduced internalization in comparison with the monolayer cells. This lack of internalization resembles the multicellular drug resistance phenomenon found in solid tumors in vivo. 31
Although both positively and negatively charged quantum dots had reduced internalization into spheroids compared to the monolayer of cells, surprisingly, negatively charged carboxyl-terminal quantum dots had significantly greater penetration into HeLa spheroids than positively charged amino-terminal quantum dots (see Figure 5). A possible explanation for this discrepancy could be the unique multiproliferative status of tumor spheroids (see Figure 2) and different transport routes of positively and negatively charged particles; that is, the positively charged nanoparticles were preferentially retained by negative surface charges on extracellular matrix and taken up by proliferating cells in the very peripheral region of the spheroids, whereas without any hindrance from the negatively charged extracellular matrix, the negatively charged nanoparticles actually diffused into the spheroids and were taken up by the viable cells. 32 Although the exact mechanisms for this charge preference remain unclear and require further experiments, in our study, using multiphoton microscopy, we demonstrated quantifiable differences between nanoparticle delivery between spheroid and monolayer cell culture systems. Furthermore, future detailed analysis using multiphoton microscopy techniques can potentially uncover the underlying mechanism between charged quantum dot delivery into cellular spheroids.
Our results of charged quantum dots differed from what were identified in an earlier study using positively and negatively charged gold nanoparticles. 32 In addition to charge, cellular penetration depends on the size, shape, and even materials of the nanoparticles. Our own unpublished results showed that with the same charge, smaller gold nanoparticles penetrated more deeply than larger gold nanoparticles; the quantum dots used in our study were 20 nm. In the earlier study, the overall size of the ligand-conjugated gold nanoparticles was only 6 nm. The larger size of our quantum dot may have contributed the difference. Another explanation is that in our study, we measured the quantum dot fluorescence directly (the fluorescence truly represents the location and amounts of the quantum dot nanoparticles); in the previous study, however, fluorescence was measured from released ligands. In the earlier study, fluorescein isothiocyanate (FITC) was conjugated to gold nanoparticles and the fluorescence of FITC was quenched; therefore, the fluorescence reappeared after FITC was released intracellularly through thiol exchange. The fluorescence reported was actually from FITC molecules that dissociated from the nanoparticles. A similar approach was implemented to conjugate doxorubicin, a molecule with inherent red fluorescence, with additional acid labile hydrazone linkage on top of thiol-Au linkage, which is cleavable in endosomes before later cleavage through thiol exchange in cytoplasm. In their study, the outcomes of doxorubicin-AuNP and FITC-AuNP were not exactly the same. Given that we used a direct approach to localize the position of quantum dot nanoparticles, (which do not dissociate), our measurements are comparable to the previous findings.
Within this study, we were also able to distinguish between different penetration behaviors of the synthetic micelles of varied compositions. PLA is a biodegradable material with low toxicity, excellent biocompatibility, and bioabsorbability. However, the low hydrophilicity and high crystallinity of PLA are limitations and need to be further improved for future drug delivery development. 33 PLGA, containing lactide/glycolide polymer chains, has greater crystallinity, permeability, and hydrophilicity than PLA homopolymers, which may explain the observed improved passage of the PLGA micelle in both monolayer cell and 3D cultures.34,35 Polymer PAL-DPPE is a derivative of polyaspartic acid. 36 Polyaspartic acid is a typical hydrophilic biodegradable polymer with excellent biocompatibility. The incorporation of DPPE, 37 a major component of the cellular inner membrane, might have enhanced the membrane permeability of polyaspartic acid and made PAL-DPPE the best performer among the three micelles tested.
Conclusion
Through the optimization of pairing SEM and TEM imaging techniques with 3D tomography multiphoton imaging, we have demonstrated that HeLa spheroids can act as a promising screening tool for various existing and novel cancer therapies, including nanoparticles.
Footnotes
Acknowledgment
Financial disclosure of authors: This work was supported by grants from the Chinese Natural Science Foundation project (No.30970784), National Key Basic Research Program of China (2009CB930200), Chinese Academy of Sciences (CAS) “Hundred Talents Program” (07165111ZX), and CAS Knowledge Innovation Program.
Financial disclosure of reviewers: None reported.