
Abstract
The multimodal strategy incorporating T1-weighted magnetic resonance imaging (MRI) and near-infrared (NIR) fluorescence imaging can complement their strengths to provide images with high sensitivity and spatial resolution for noninvasively and dynamically monitoring endothelial progenitor cells (EPCs) in potential EPC-dominated therapies. Here we report the development of a protein-based imaging probe, bCD-PLL-Cy5.5 Conjugate 1, in which the bacterial cytosine deaminase (bCD) protein was modified with poly-
WITH ITS NONINVASIVE CHARACTERISTICS and exquisite spatial resolution, magnetic resonance imaging (MRI) is one of the most powerful techniques available for imaging diagnosis, and preclinical results can be relatively easily translated to clinical applications. 1 However, MRI suffers from suboptimal detection sensitivity. 2 Optical imaging, on the other hand, is highly sensitive and capable of detecting a small amount of light-emitting materials, but its image formation is unsatisfactory because of the intrinsic absorption and light scattering of heterogeneous tissues,1,3 and it is not readily transferable to the clinic. In preclinical studies, a multimodal strategy incorporating MRI and optical imaging can complement their strengths while minimizing their weaknesses.
Iron oxide nanoparticle–based MRI contrast agents effectively generate a decreased signal on T2-weighted images. However, a considerable disadvantage of this T2 effect is that magnetic field inhomogeneities can be mistaken for a contrast agent effect. In addition, these contrast agents are metabolized intracellularly, leading to a slowly decreasing contrast effect over time 4 and possible toxicity owing to accumulation of nonphysiologic intracellular iron. 5 Especially after interventional procedures for stem cell delivery, the signal loss induced by iron oxide nanoparticle–based contrast agents may be difficult to distinguish from the susceptibility artifacts from air or postsurgical iron or hemosiderin deposition. 6
Despite having lower detection sensitivity compared to iron oxide nanoparticle–based contrast agents, T1 contrast agents are advantageous for enabling a positive contrast on T1-weighted images, which is less likely to be confused with artifacts owing to metal- or air-induced postoperative local signal voids. Gadolinium (Gd)-based contrast agents such as gadolinium-tetraazacyclododecanetetraacetic acid (Gd-DOTA) and gadophrin-2 have been successfully applied in previous cell labeling and in vivo cell-tracking studies.7,8
Endothelial progenitor cells (EPCs) are the precursors of vascular endothelial cells. They play important roles in postnatal neovascularization and in maintaining vascular homeostasis in adults, that is, vasculogenesis and angiogenesis.9–10 EPCs can be used as new seed cells for cell treatment, as well as the new carrier cells for gene therapy. Three areas of therapeutic applications of EPCs include the repair of injured vessel walls, the neovascularization or regeneration of ischemic tissue, and the coating of vascular grafts. 9 Studies have shown that EPCs promote tissue vascular regeneration when introduced in vivo and thus provide potential therapies for cardiac ischemia, vascular insufficiency, wound healing, and tumor inhibition. 11 Clinical studies using EPCs for neovascularization have just started.12,13 However, the mechanisms for stimulating or inhibiting the differentiation of EPCs in vivo and the signals causing their migration and homing to sites of injured endothelium or extravascular tissue are largely unknown at present. On the other hand, identifying the transplanted donor cells and monitoring their viability in vivo are essential for better understanding of their migration and differentiation. We selected this cell type as the target when carrying out the magnetic or fluorescent labeling of stem cells.
In this study, the bacterial cytosine deaminase (bCD) protein and the poly-
Materials and Methods
Synthesis of bCD-PLL Conjugate 1
bCD-PLL was prepared according to the synthetic procedure as previously described, 15 and the structure is shown in Figure 1. Briefly, bCD protein was isolated from transformed Escherichia coli cultures. Gd3+-DOTA chelators, fluorescent probe including rhodamine, and Cy5.5 were functionalized into the PLL moiety. The conjugation between bCD and PLL labeled with imaging reporter forms the aiming probe.

The structure of Conjugate 1.
Isolation of Peripheral Blood Mononuclear Cells
The study was approved by the institutional Animal Use and Care Committee. EPCs were isolated from newly collected rabbit blood. A New Zealand white rabbit weighing 2.5 kg (Jingling Farm Center for Animal Experiments, Nanjing, China) was anesthetized intravenously with 10 mg/kg pentobarbital sodium. Fresh blood (20 mL) was collected by a direct puncture into the left ventricle with a 14-gauge needle. The blood was heparinized (100 IU/mL heparin sodium, Qianhong Inc., Jiangsu, China), diluted to a 1:1 ratio with phosphate-buffered saline (PBS), and centrifuged in a density-gradient centrifuge Histo-Paque 1077 (Sigma-Aldrich, St. Louis, MO) at 400g for 30 minutes. A sedimented layer of peripheral blood mononuclear cells was collected, washed twice with PBS, adjusted to a concentration of 5 × 105/mL, and suspended in 5 mL of microvascular growth medium 2 (EGM-2 MV, Cambrex, Walkersville, MD). The cells were placed in a 25 mm2 culture flask (Corning Inc., Corning, NY) and cultured at 37°C with 5% CO2. Four days later, the culture solution was replaced for the first time, and the cells not adhering to the flask wall were removed. Later, the cultured solution was replaced every 3 days until the number of cells adhering to the flask wall reached about 90% area of the flask wall. The original medium was discarded; 1.5 mL of 0.25% trypsin was added to the flask to separate the cells from the flask bottom. After fresh culture medium was added again, the cells were passed in 1:2 and marked as passage 1 (P1). During the passage, the culture medium was replaced every 3 days until the cells were grown to confluence in the culture flask. The procedure above was repeated and the cells were marked as passage 2 (P2), passage 3 (P3), and so on. Cells were passaged up to nine times after reaching confluence. To ensure the healthy growth of cells in the culture, cell morphology was checked on a daily basis with an inverted phase-contrast microscopy (Axioscop, Zeiss Co. Ltd., Oberkochen, Germany).
To characterize the cultured EPCs, their capacity to uptake DiI-labeled acetylated low-density lipoprotein (DiI-Ac-LDL) and ability to bind to fluorescein isothiocyanate (FITC)-labeled Ulex europaeus agglutinin–1 (FITC-UEA–1), which is a function of endothelial cells, were tested. The adherent cells that stained positive with both FITC-UEA-1 and DiI-Ac-LDL were indicated to be differentiating endothelial cells. 16
For cellular labeling, cells grown in 60 mm plates were incubated with Conjugate 1 for 24 hours. At the end of treatment, the cells were washed with PBS three times to remove the excess Conjugate 1, and labeled cells were identified with fluorescence microscopy.
Confocal Laser-Scanning Fluorescence Microscopy
All fluorescence microscopic images were performed with a Zeiss LSM 510 META confocal laser-scanning microscope (Carl Zeiss, Inc., Oberkochen, Germany) using a Plan-Apochromat 63 × /1.4 oil immersion lens (Zeiss). All labeled cells were excited with a HeNe laser (543 nm), and fluorescence of rhodamine was detected by a secondary photomultiplier by applying a 575 to 650 nm band-pass filter.
Flow Cytometric Analysis
For the studies of cell uptake kinetics and concentration-dependent internalization, cells grown in 60 mm plates were incubated with 2 μM of Conjugate 1 for 24 hours. At the end of treatment, the cells were washed with PBS three times to remove the excess Conjugate 1, trypsinized, and then washed again with PBS. After centrifugation, the cell pellet was resuspended in 0.6 mL of 0.5% paraformaldehyde and stored at 4°C until analysis. Flow cytometry was performed using a FACS Calibur instrument (Becton Dickinson, San Jose, CA) equipped with a 633 nm HeNe laser. Then 5,000 events per sample were analyzed. CellQuest 3.3 software (BD Bioscience, San Jose, CA) was used for data acquisition and analysis.
Effects of Conjugate 1 Labeling on Cell Proliferation and Cell Cycling
3-(4,5-Dimethylthiazol-2-Yl)-2,5-diphenyltetrazolium bromide (MTT) assays were performed for assessment of toxicity and normal proliferation of labeled cells. The EPCs of P3 were grown in 72 wells of a 96-well plate at 104 cells per well, and the Conjugate 1 solution was added to the wells at final concentrations of 0.25, 0.5, 1, 2, and 3 μM. Twelve duplicated wells were set for each concentration, and the other 12 wells were kept free from Conjugate 1. On days 1 and 5, after the culture medium containing Conjugate 1 was discarded, the cells were washed with PBS. After continuous culture in 5 days, 20 μL of MTT solution (5 mg/mL, Fluka Co., Buchs, Switzerland) was added to each well and then incubated for 4 hours at 37°C in a 5% CO2 atmosphere. After supernatant was discarded, 150 μL of dimethylsulfoxide (Shanghai Bioengineering Co., Shanghai, China) was added to each well, shaking for 10 minutes. Light absorption values of each well were measured at 570 nm using a spectrophotometer (Sepctra MAX 250, Molecular Devices Corp, Sunnyvale, CA).
The cell cycle analysis was performed with a flow cytometer (Becton, Dickinson, San Jose, CA) in 5 × 105 cells (unlabeled and labeled with 2 μM of Conjugate 1) after they were fixed in 70% ethanol at 4°C for 24 hours and incubated with 5 μL (10 μg/mL) ribonuclease inhibitor at 37°C for 30 minutes and 5 μL (50 μg/mL) of propidium iodide at 4°C for 5 minutes.
MRI Phantom Preparation
At 24 hours after incubation, cells were washed twice with PBS, harvested using trypsin, and counted. For the cell phantom, 1 × 106 labeled and unlabeled cells were resuspended in 0.5 mL of 1% agarose in Eppendorf tubes of 0.5 cm in diameter, respectively. Another Eppendorf tube was used to contain distilled water.
MRI and Data Acquisition
All MRI experiments were performed on a 7 T scanner with a 31 cm diameter horizontal bore magnet (Bruker PharmaScan, Ettlingen, Germany). The horizontal bore system is equipped with a 15 cm diameter gradient set capable of generating 740 mT/m gradient strengths in all three directions. A 23 mm diameter circular volume coil was used for MRI data collection.
The MRI sequences were a multislice T1-weighted spin-echo (repetition time ms/echo time ms, 600/7.5) and a multislice T2-weighted fast spin-echo (repetition time ms/echo time ms, 4200/36) sequence. Images were obtained using a section thickness of 0.5 mm, a field of view of 3 × 3 cm, and a matrix size of 192 × 512 with two measurements. A round region of interest of 10 mm2 was used for signal intensity measurement. Signal to noise ratios (SNRs) were calculated by dividing the signal intensity of region of interest by the background noise of the image, which was measured in the background anterior to the depicted object.
The T1 relaxation time and longitudinal relaxivity, r1 of cell pellets treated or without the treatment of Conjugate 1 was determined at the field strength of 7.0 T at 25°C. An inversion recovery sequence (repetition time = 3,000 seconds) was used with 10 different recovery times (ie, 5, 10, 20, 40, 60, 80, 100, 200, 400, 1,000 ms). A nonlinear fitting algorithm with the exponential recovery function was used to fit the observed magnetic resonance signal intensities at different recovery times to give the T1 relaxation time for each particular sample. Four concentrations were used to determine the r1 value by plotting the reciprocals of T1 against the concentration of the Gd3+ ions according to the equation r1 = (1/Tip −1/T1d)/[Gd], where r1 is the T1 weighted relaxivity, T1p is the paramagnetic T1 value, T1d is the diamagnetic T1 value, and [Gd] is the gadolinium concentration.
In Vitro Optical Imaging Studies
Optical images were obtained with a Kodak In-Vivo Imaging System FX Pro (Rochester, NY). DsRed filter sets (excitation/emission: 550–555 nm/575–650 nm) were used for acquiring rhodamine images, whereas Cy5.5 filter sets (excitation/emission: 675 nm/695 nm) were used for acquiring Cy5.5 images. All fluorescent images were acquired using 0.1-second exposure time (field of view 12.8 cm; f/stop 4; no binning), and the fluorescence intensity was scaled as a unit of ps−1 cm−2 sr−1. A series of images were acquired. White light images were acquired before acquisition of each fluorescent image using the same field of view. Data were analyzed with Kodak Molecular Imaging System software, and coregistered white light and fluorescent images were generated using Photoshop 7.0 software (Adobe).
Statistical Analysis
Statistical analyses were performed with SPSS for Windows software version 11.0 (SPSS Inc, Chicago, IL). Numeric data including SNRs, MTT absorbance value, and cell cycle count were reported as means ± standard deviation. For statistical comparisons, three repeated measures analysis of variance was used. A p value of less than .05 was considered to indicate a significant difference. The differences of MTT mean values between the labeled and unlabeled EPCs at each concentration and each time point were subjected to the Kruskal-Wallis rank sum test, whereas the differences of mean SNRs and cell cycle count values between the groups were subjected to the Student t-test.
Results
Specifications of Conjugate 1
The molar ratio of bCD hexamer/PLL/rhodamine/Cy5.5/Gd3+-DOTA/biotin in Conjugate 1 was measured as 1:1:1:1:15:3. The molecular weight of Conjugate 1 was determined by size-exclusion chromatography. The superimposed peaks monitored at 280 nm (bCD protein) and 635 nm (Cy5.5) confirmed covalent conjugation between PLL and bCD (data not shown). The molecular weight of Conjugate 1 was measured as 345 kDa. The mean hydrodynamic radius of Conjugate 1 was determined as 24.4 nm. The conjugation of rhodamine to PLL resulted in a slight red shift of the emission spectrum centered at 590 nm. At 7.0 T, 25°C, the water proton longitudinal relaxivities r1 of Conjugate 1 and Gd-DOTA as a control contrast agent were measured as 8.6 and 4.2 mM−1 s−1/Gd3+ ion, respectively, at pH 7.4.
Morphologic Observations of Living Cells
Under inverse microscopy, freshly isolated EPCs appeared as round cells with variable sizes (Figure 2A). After 4 days in culture, the cells became spindled, polygonal, or triangular in shape with a single, centrally located nucleus and tended to form cluster-like colonies (Figure 2B). The cells divided rapidly, changing rapidly from packed round cells into a contact-inhibited monolayer (Figure 2C). On day 13 in culture, the clustered cells were seen to be in contact with each other and grew into the confluent monolayer, which was mostly completed by day 18 in culture (Figure 2D). The cultured EPCs showed the typical “cobblestone” appearance of endothelial cells. EPCs were further identified from internalization of DiI-Ac-LDL and binding to FITC-UEA-1. 17
Confocal Laser-Scanning Fluorescence Microscopy
Cellular uptake of Conjugate 1 was investigated in EPCs. A concentration of 2 μM of Conjugate 1 was shown to label EPCs efficiently without causing alterations in cell morphology and proliferation. Figure 2E shows fluorescence images of cells after treatment with 2 μM of Conjugate 1 for 24 hours at 37°C. Fluorescence of both rhodamine and Cy5.5 was detected after being excited at 530 and 640 nm, respectively (Figure 3). Clear and well-matched vesicular structures are predominantly located in the perinuclear region of cells. The strong intracellular fluorescence signal is consistent with the flow cytometry results.
Kinetics of Internalization of Conjugate 1
The cellular internalization kinetics of Conjugate 1 in EPCs is shown in Figure 4A. Conjugate 1 (2 μM) in the serum-free medium was added to the cells, and the cellular fluorescence was quantitatively assessed at selected time points by flow cytometry after washing with trypsin to remove any nonspecific bound conjugate on the cell surface. Analysis of the flow cytometry profiles (upper profile of Figure 4) and time-dependent cellular fluorescence values (Figure 5) clearly indicates that the cellular uptake of Conjugate 1 rapidly increased in a nearly linear manner during the first hour, followed by a slow and progressive increase and an apparent plateau from 24 to 48 hours. Meanwhile, the symmetric and narrow cellular fluorescence profiles shown in Figure 4 suggest a homogeneous uptake of Conjugate 1 by cells.
MTT Test and Cell Cycle
The MTT absorbance values of EPCs labeled with different concentrations of Conjugate 1 (0.25, 0.5, 1, 2, and 3 μM), when compared to unlabeled EPCs on the first and fifth day, also show no significant difference between the labeled and unlabeled EPCs at each concentration and each time point (Kruskal-Wallis rank sum test) (see Figure 5).
The flow cytometer assay of the cell cycle reveals that 92% of cells were in the G0/G1 phase, whereas the rest were in the (S + G2 + M) phase. There is no significant difference between unlabeled and labeled cells: 92.01 ± 0.06% versus 91.27 ± 0.04% (p > .05) (Figure 6).

Phase-contrast microscopy of the morphologic changes in New Zealand white rabbit blood endothelial progenitor cells (EPCs) (A–D). A, Freshly isolated EPCs appeared as round but anisodiametric cells (X200 original magnification). B, EPCs tended to form cluster-like colonies, as seen on the fourth day of culture (X 200 original magnification). C, The cells divided rapidly, from packed round cells into a contact inhibited monolayer (X200 original magnification). D, On day 18 of culture, EPCs showed endothelium-like cobblestone morphology (X400 original magnification). E, Fluorescence image of labeled cells (X 400 original magnification).
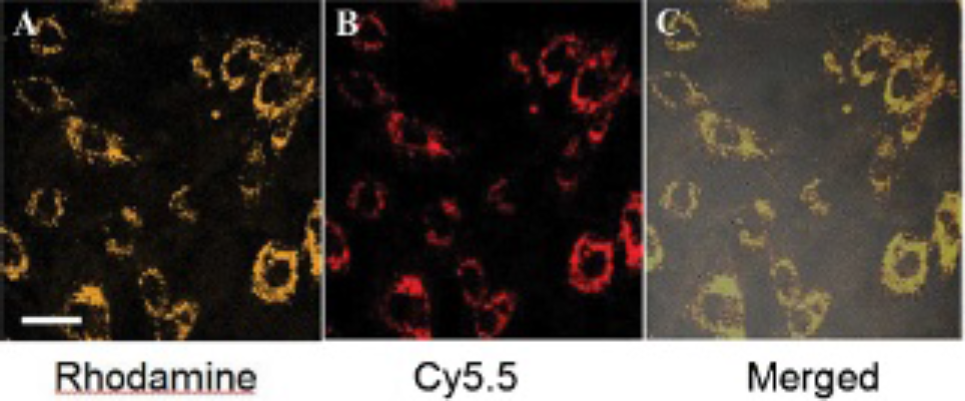
A to C demonstrate the rhodamine (A), Cy5.5 (B), and merged confocal fluorescence (C) images of live endothelial progenitor cells treated with 2 μM of Conjugate 1 for 24 hours. The scale bar presents 15 μm.
Magnetic Resonance Imaging
The MRIs by T1-weighted magnetic resonance sequences from 1 × 106 labeled and unlabeled EPCs of 24 hours' culture are shown in Figure 7. T1-weighted images demonstrated a significant increase in SNRs for labeled (1,980 ± 182) compared to unlabeled cells (960 ± 50, p < .05). There was no significant signal intensity decrease on T2-weighted images.
Calculating from in vitro T1 maps of cell pellets, T1 values of 1 × 106 labeled and unlabeled cells are 340 and 1,080 ms, respectively. Conjugate 1 labeling shortened the cells' T1 relaxation time (see Figure 7).
In Vitro Optical Imaging Studies
Figure 8 shows the NIR optical imaging of the cell pellets of EPCs (see Figure 8A) treated with 2 μM of Conjugate 1 for 24 hours.

Kinetics of internalization of Conjugate 1 in endothelial progenitor cells. The cells were incubated for indicated periods of time, and then cell fluorescence was analyzed by flow cytometry. The lowest peak shows autofluorescence of cells before the treatment. bCD-PLL = bacterial cytosine deaminase protein and the poly-
Discussion
Iron oxide nanoparticles have been in use for several years as an effective T2/T*2 contrast agent for labeling and tracking transplanted stem cells. Magnetic resonance tracking studies have been performed in the brain, 18 spinal cord, 19 myocardium, 20 liver, 21 and kidney. 22 However, in many cases, it is difficult to distinguish labeled cells from other hypointense regions on T2/T*2-weighted MRIs. These hypointensities can have a physiologic origin, such as hemoglobin in blood, or a pathologic origin, such as blood clots or air from experimental, traumatic procedures. One attempt to differentiate iron-labeled cells from blood vessels was to alter the inhaled oxygen levels to reduce the blood oxygenation level-dependent (BOLD) effect. 23 Nevertheless, hypointensities on MRIs remain a major obstacle for increased specificity of cell tracking, preventing this method from being used in certain applications, particularly those that involve trauma and hemorrhage. In addition, these contrast agents are metabolized intracellularly, leading to a slowly decreasing contrast agent effect over time 4 and possible toxicity owing to nonphysiologic intracellular iron concentrations. 5 Therefore, an alternative method of detecting cells is to use contrast agents that create bright contrast or hyperintensity.
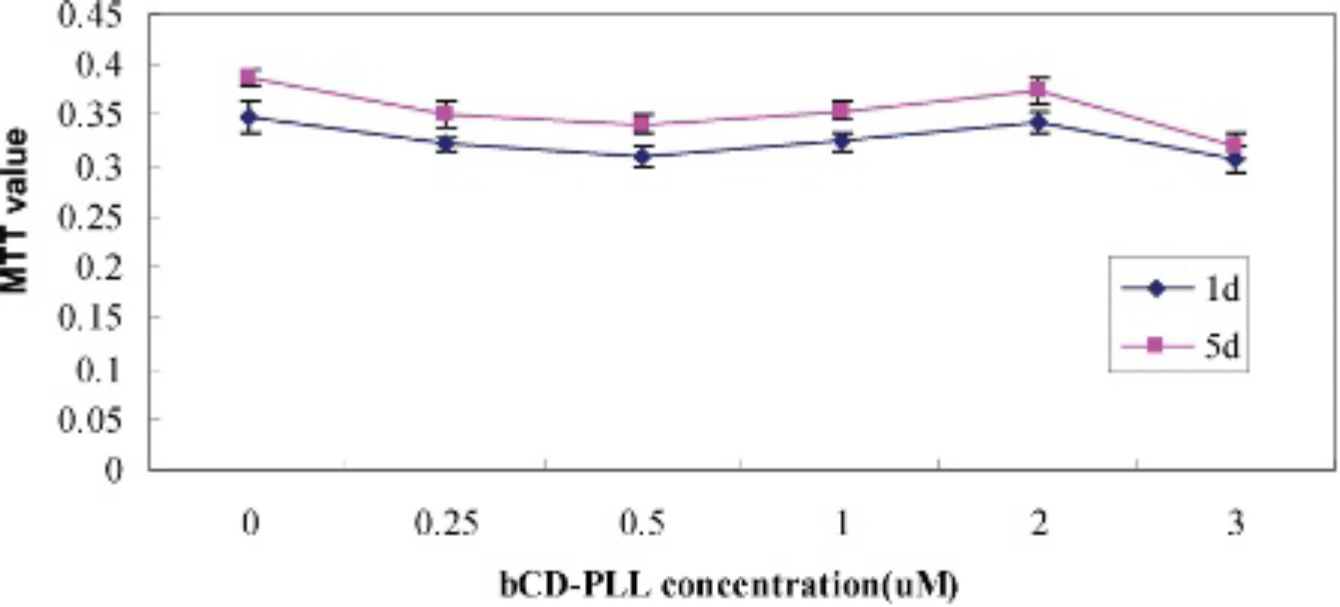
Relative cell viability of endothelial progenitor cells at days 1 and 5 after incubation with different concentrations of Conjugate 1 for 24 hour. There are no statistical differences in MTT values. bCD-PLL = bacterial cytosine deaminase protein and the poly-
Other than iron oxide nanoparticle labels, Gd complexes have been investigated for labeling fibroblasts with direct gadolinium diethylenetriamine pentaacetic acid (Gd-DTPA) bovine serum albumin incubation 24 or incubation with Gd-DTPA/fatty acid complex.14,25 In these cases, “positive” contrast can be observed. Moreover, positive Gd-based T1 contrast agents can provide better anatomic details on MRIs of T1-weighted sequence. 14 Depending on the specific application, labeling cells with Gd-conjugate nanoparticles could be a viable alternative for labeling of iron oxide nanoparticles. These applications may include experimental settings where it is difficult to distinguish iron oxide nanoparticle–labeled cells from blood/hemoglobin-derived hypointense regions or from tissues and organs that have inherently high concentrations of iron, such as the liver, or certain tumors, such as melanomas. But it is well known that the major shortcoming of Gd-conjugate is lower T1 relaxivity when compared to iron oxide nanoparticles.

Cell cycle analyses show that the unlabeled and labeled endothelial progenitor cells in the G0/G1 state at day 1 were 91.30% and 92.07%, respectively.
In this work, we developed a novel multimodal imaging reporter Conjugate 1 of positive magnetic resonance contrast with high T1 relaxivity and strong fluorescence signals. In contrast to the negative contrast from iron oxide nanoparticles on T2-weighted MRIs, here we explored the use of a positive contrast agent that can provide opposite contrast on the generated image using a newly developed nanoparticle preparation. bCD is an enzyme exploited in cancer gene therapy owing to its high stability. 26 PLL was selected as a carrier of the imaging reporters because it is biodegradable and its extended random coil conformation may facilitate the extravasation of the conjugate into the interstitium. 27 PLL was functionalized with Gd3+-DOTA, rhodamine, and Cy5.5 for dynamically monitoring the distribution of bCD in vivo by either MRI or optical imaging. Rhodamine can be used to track the probe in the excised tissue; Cy5.5 can be used for in vivo NIR fluorescence imaging. Thus, we intended to optimize Conjugate 1 with improved biocompatibility for biomedical applications.

A to C present the T1 weighted images of endothelial progenitor cell pellets treated with 2 μM of Conjugate 1 for 24 hours (A), without any treatment (B), and with double distilled water (C). D to F present the T1 map of the cell pellets shown in A to C.

A to D present the near-infrared optical imaging of the cell pellets of endothelial progenitor cells (EPCs) (A, C) treated with 2 μM of Conjugate 1 for 24 hours. B and D show the EPC pellets without the treatment. A and B are images with excitation/emission of 550 to 555 nm/575 to 650 nm filter sets acquiring rhodamine images, whereas C and D are images with excitation/emission of 675 nm/695 nm filter sets acquiring images with Cy5.5. Top arrows present the central area of cell pellets, whereas bottom arrows present the peripheral area of cell pellets.
At 7.0 T and 25°C, the water proton longitudinal relaxivities rlp of Conjugate 1 were measured as 8.6 mM−1 s−1/Gd3+ ion at pH 7.4, 15 superior to clinically used MRI contrast agents such as Gd3+-DOTA (r1p = 4.3 mM−1 s−1). This may diminish the drawback of the limited sensitivity of Gd-based contrast agents when compared to iron oxide-based contrast agents. On the other hand, Conjugate 1 is an amphiphilic molecule and thus is able to dissolve in aqueous solutions and plasma. The amphiphilic structure allows the Gd molecule to penetrate phospholipid bilayers 28 and to interact with intracellular water protons, which increases T1 relaxation times and leads to the observed increased signal on T1-weighted images. In this study, when Conjugate 1 was labeled into EPCs, the T1 value of 106 EPCs was three times shortened, from 1,080 ms for unlabeled cells to 340 ms for labeled cells. By comparison, conventional Gd-chelates are highly hydrophilic and thus are not taken up spontaneously by cells.
Interestingly, the cellular T1 relaxivities were different for the cell pellet and cells suspended in gelatin (340 ms and 10 ms, respectively), indicating that the microenvironment of the cells has considerable impact on the relaxivities. This is also true in vivo in cell transplantation, in which the relaxation time measured from each voxel is composed not only from the transplanted cells but also from a variety of inhabitant cells and the extracellular matrix. Although we do not have direct evidence to explain possible mechanisms, we are aiming to elucidate this in the future.
The uptake of contrast agent by monocytes could have occurred via three mechanisms: (1) endocytosis, (2) transmembrane transport, or (3) intercalation and cell membrane turnover. We have demonstrated that the cellular uptake of Conjugate 1 is an energy-dependent process that is mediated in part by the character of the PLL moiety but is independent of serum components. 29 PLL moiety may facilitate the cell uptake of Conjugate 1 through polycation-mediated endocytosis. 30 Interestingly, NIR fluorescence imaging of the equal number of cells at the same time point showed a higher fluorescence intensity of labeled EPCs than that of human umbilical vein endothelial cell line ECV304 (data not shown). This variability may be caused by the different phagocytic activities of the various cell cultures, size of the membrane surface, and charge.
After 24 hours' incubation, the surviving cells showed viability similar to that of controls, as manifested by MTT and the cell cycle. It is well known that free Gd can be toxic to mammalian cells. In this study, we used 2 μM of Conjugate 1 to achieve a maximal loading of the cells. However, it has no observable cellular toxicity even at this high concentration.
Optical imaging is an emerging noninvasive diagnostic modality that offers advantages including nonionizing radiation, high sensitivity, low cost, and the possibility of real-time image-guided surgical procedures31,32 compared to conventional techniques, such as MRI and positron emission tomography. For future applications of clinical optical imaging, probes that are emissive in the NIR region of 700 to 900 nm are currently being developed.33–37 The NIR region is advantageous for in vivo optical imaging because the background from tissue autofluorescence and absorption from intrinsic chromophores are low, allowing NIR light to penetrate several centimeters into heterogeneous tissues. 3 NIR imaging has shown clinical potential especially in breast imaging because of convenience for direct contact of breast tissue with NIR sources and detectors. Several clinical trials are currently ongoing.38,39
We synthesized the multimodal probe with two fluorophores and evaluated their labeling capability in vitro. The optical imaging signal from Conjugate 1 was improved by combining rhodamine and NIR fluorophore Cy5.5. The advantages of using NIR fluorophores for in vivo optical imaging are the low tissue autofluorescence and absorption from intrinsic chromophores in this region. The probe can also potentially be used for in vivo cellular imaging. Thus, by using this multimodal contrast agent, transplanted cells may be identified by MRI and NIR imaging for a series of evaluation in vivo and by postmortem fluorescent microscopy to validate the in vivo observations. The ability to image implanted stem cells in vivo will further our understanding of the role of cell transplantation, provide a potential means of diagnosis, and help in developing cell therapies.
Our experiments showed that EPCs incubated ex vivo with Conjugate 1 spontaneously internalized this particulate, soluble, macromolecular contrast agent. Cellular uptake after incubation with this conjugate was sufficient for detection of the cells by both T1-weighted MRI and optical imaging.
In summary, we have developed a novel multimodal imaging probe, Conjugate 1, demonstrating low cytotoxicity, efficient cell uptake, high T1 relaxivity, and a strong fluorescence signal in EPCs. We have shown in vitro Conjugate 1 as an MRI and optical imaging “double-labeling” probe with multimodality contrast. In vivo studies of this conjugate in cellular tracking in tumor xenograft models are currently under way.
Footnotes
Acknowledgments
Financial disclosure of authors: Supported by the National Nature Science Foundation of China (No.30830039, No. 30900353) and Major State Basic Research Development Program of China (973 Program) (No. 2010CB933903).
Financial disclosure of reviewers: None reported.