
Abstract
During the last decade, there has been enormous progress in understanding both multipotent stem cells such as hematopoietic stem cells and pluripotent stem cells such as embryonic stem cells and induced pluripotent stem cells. However, it has been challenging to study developmental potentials of these stem cells because they reside in complex cellular environments and aspects of their distribution, migration, engraftment, survival, proliferation, and differentiation often could not be sufficiently elucidated based on limited snapshot images of location or environment or molecular markers. Therefore, reliable imaging methods to monitor or track the fate of the stem cells are highly desirable. Both short-term and more permanent monitoring of stem cells in cultures and in live organisms have benefited from recently developed imaging approaches that are designed to investigate cell behavior and function. Confocal and multiphoton microscopy, time-lapse imaging technology, and series of noninvasive imaging technologies enable us to investigate cell behavior in the context of a live organism. In turn, the knowledge gained has brought our understanding of stem cell biology to a new level. In this review, we discuss the application of current imaging modalities for research of hematopoietic stem cells and pluripotent stem cells and the challenges ahead.
Unique Biology of Hematopoietic Stem Cells and Pluripotent Stem Cells
Recent advances in stem cell research have provided unprecedented opportunities for cell replacement therapies. However, much has yet to be learned to more efficiently isolate, expand, and manipulate the fate of stem cells for clinical purposes. How stem cells develop in vivo (in the body of a live organism) and how the microenvironment affects stem cell development are among the key questions that need to be addressed. As we move forward to therapeutic approaches, it is also essential to demonstrate the safety and the efficacy of stem cell transplantation before use in patients. Among many technologies developed for modern biomedical research, cell imaging has been instrumental for these purposes and will continue to be indispensable for stem cell research.
The hematopoietic stem cell (HSC) has served as a model system for studying stem cell biology ever since the first reports of the existence of self-renewing populations.1,2 HSCs give rise to all the blood and immune cells in the body. Studying HSCs has led to the most common and successful clinical application of stem cells, bone marrow transplantation (BMT). However, wider applications of BMT have been limited by the limited supply of available HSCs that are genetically and immunologically compatible with potential recipients. 3 This reflects our current lack of understanding of HSC biology that is essential for stem cell isolation and expansion. HSCs exist at a very low frequency and mostly remain quiescent in their native bone marrow niche, making stem cell selection extremely difficult.4,5 Ex vivo expansion (ie, in the laboratory mimicking in vivo conditions) of HSCs requires a comprehensive understanding of the molecular events and environmental cues regulating HSC self-renewal and differentiation. Many hematologic disorders may also have defects within their HSC microenvironment 6 ; therefore, effective treatment of these types of diseases also depends on the understanding of mechanisms underlying the stem cell niche regulatory functions. Although the HSC niche has been studied extensively, the bone marrow niche remains a complex environment consisting of many different types of cells and extracellular matrix components, which makes direct visualization of HSCs and HSC-niche interactions difficult. It is therefore important to develop proper cell labeling technologies to mark HSCs and niche cells that are suitable for imaging.
A more pluripotent stem cell, the human embryonic stem (hES) cell, was derived from the blastocyst stage of embryos by isolating the inner cell mass and plating these cells onto mouse embryonic fibroblast-coated plates. 7 hES cell lines can be maintained in culture for extended periods of time and most importantly have the ability to retain both normal karyotypes and pluripotency (ie, the potential to differentiate into cell types originated from all three embryonic germ layers) after prolonged culture. 8 For these reasons, hES cells have been anticipated as an alternative source to generate abundant cells resembling adult stem cells (eg, HSCs) for basic research purposes and for future cell replacement therapy. The recent discovery of induced pluripotent stem (iPS) cells has made it possible to generate patient-specific pluripotent stem cells from adult somatic cells.9,10 Although this research field is still in its infancy, it has been shown that iPS cells possess pluripotency with the ability to differentiate into various somatic cells (similar to hES cells), making personalized stem cell therapy a possibility.9–13 To investigate the efficacy and safety of hES/iPS-derived cells in transplantation settings, one has to have the ability to track and observe the transplanted cells in recipients over time.
Stem Cell Labeling and Imaging Technology
In certain cases, bright field imaging can give sufficient information regarding particular cellular events such as proliferation or differentiation, especially under laboratory cell culture settings, where cell numbers are low and cell layers are not deep. However, in most in vivo or ex vivo conditions, stem cells are in direct contact with an enormous number of other cell types, and it is necessary to distinguish them from their environment by cell labeling. When tissues or organs are dissected out of host animals, staining with various dye-conjugated antibodies and detection with fluorescent or light microscopy can provide high-resolution snapshots of the stem cell migration, engraftment, proliferation, and differentiation in both the short (ie, days) and long term (ie, months) after transplantation.14–16 We have used this technology to locate human HSCs after short-term migration via transplantation into mouse bone marrow (Figure 1, A-C) as well as long-term engraftment of hES cell-derived hepatocytes in mouse liver (Figure 1D). In addition, a series of pretransplantation labeling techniques including fluorescent dyes or proteins, bromodeoxyuridine (BrdU, a traceable thymidine analogue marking progeny cells after deoxyribonucleic acid [DNA] replication), bioluminescence, and magnetic labeling have all been used for tracing stem cells.17–24 Choices of labeling methods and imaging techniques are not random and should be made based on the purpose and requirement of the experiment, especially the desired resolution and duration of the studies (Table 1).

Detection of human stem cell migration and engraftment in immunodeficient mice using immunolabeling methods with specific antibodies. A to C, Short-term distribution of human hematopoietic stem cells (HSCs) in mouse marrow 2 days after transplantation into NOD/SCID/IL-2Rγ–/– (NSG) mice. A, A large-scan image of mouse femur metaphysis after staining for human HSCs with a human-specific CD45 antibody (in green). Cellular nuclei were stained by DAPI (blue). B, An enlarged image of the selected area in the red box as shown in A. Some human HSCs are in contact with trabecular bone (TB) (20× objectives). C, A 100× confocal image of the same marrow section after costaining for a preosteoblast marker, N-cadherin (red), and a human HSC marker, CD34 (green). Some CD34+ HSCs are in contact with preosteoblasts lining the endosteum of TB. D, Long-term engraftment of human embryonic stem (H1)-derived mature hepatocytes. NSG mouse liver was harvested at 2 months post-transplantation of 0.1 million hepatocytes differentiated from human embryonic stem cells in a defined culture condition. 49 Human hepatocytes that are producing the albumin protein (shown in green) are identified by an antibody specifically recognizing human but not mouse albumin. A control mouse liver that was not injected with human cells is shown in the left corner of D, and no positive cells were detected. Both marrow and liver images were taken using the motorized Nikon Ti-E microscope with a Nikon Encoded Motorized XY stage and a function in NIS-Elements – Advanced Research software (Nikon, Melville, NY) called “Scan Large Image” to generate these montaged images. The camera used is a Coolsnap HQ2 (Photometrics, Tucson, AZ).
Comparison of Different Labeling Methods in Stem Cell Research
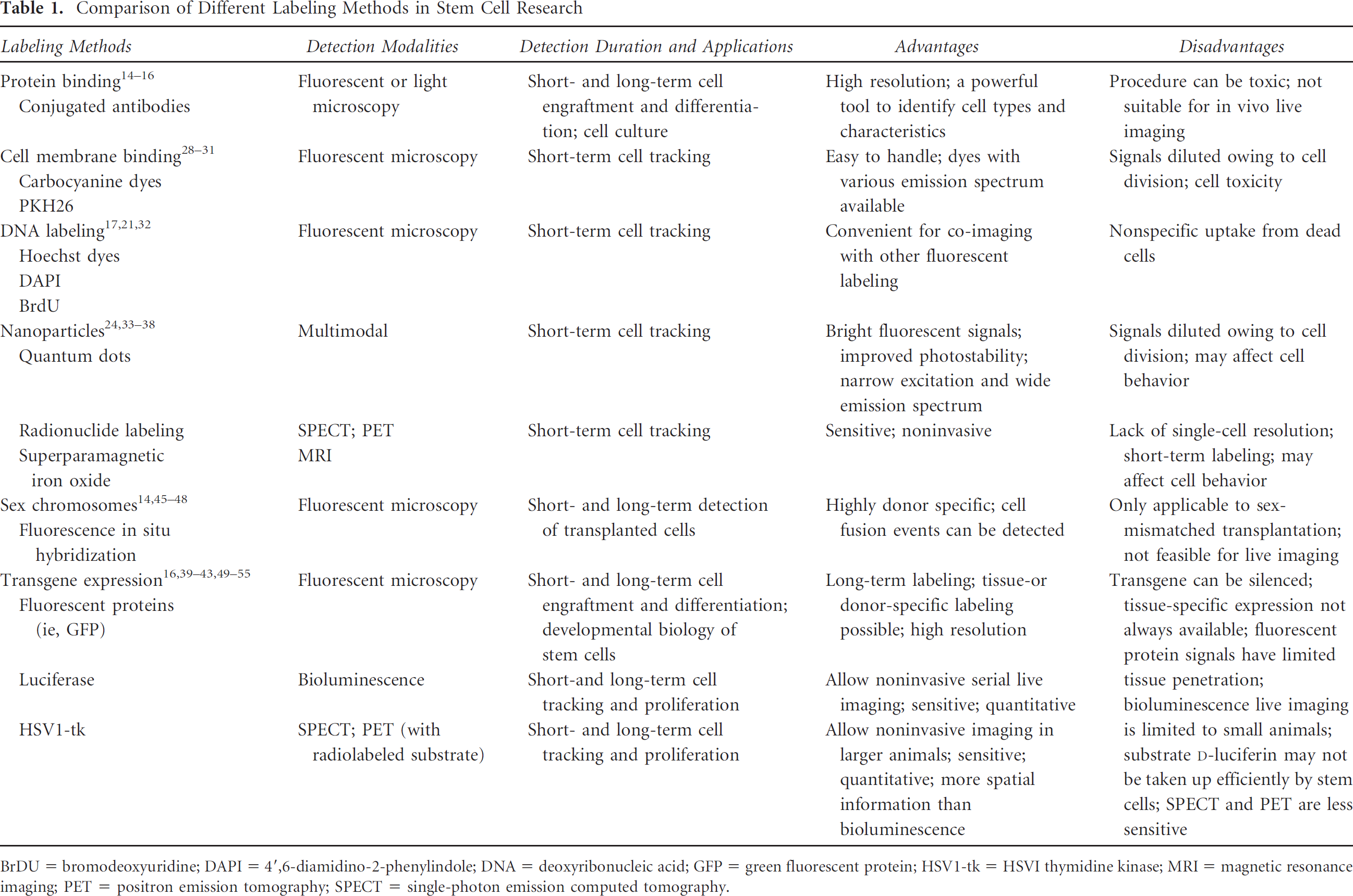
BrDU = bromodeoxyuridine; DAPI = 4′,6-diamidino-2-phenylindole; DNA = deoxyribonucleic acid; GFP = green fluorescent protein; HSV1-tk = HSVI thymidine kinase; MRI = magnetic resonance imaging; PET = positron emission tomography; SPECT = single-photon emission computed tomography.
Fluorescent labeling by either chemical dyes or proteins tends to give higher single-cell sensitivity and resolution when monitored with fluorescent microscopes. When imaging cells in relatively deeper specimens, confocal microscopy has been a conventional choice because it uses a unique pinhole to exclude out-of-focus background fluorescence from detection, allowing three-dimensional sectioning of thicker tissues. Recent development of multiphoton microscopy offers additional advantages for such purposes.25,26 In a two-photon laser scanning microscope, the excitation is generated only at the focal point. Owing to the relatively low energy of each individual photon, autofluorescence and autophoto-bleaching at out-of-focus areas are significantly reduced; therefore, the specimen penetration is also significantly increased over traditional confocal microscopy. The reduced damage to cells across the imaging paths also makes this technology a better alternative for imaging live tissues.
Bioluminescence or magnetic resonance imaging (MRI) modalities in general lack single-cell resolution in comparison to fluorescence imaging. On the other hand, whereas fluorescence imaging in animals often requires exposing the tissue or organ during the imaging process, bioluminescence and magnetic resonance retain higher tissue-penetrating abilities 27 and therefore are more suitable for noninvasive live imaging. Unlike fluorescence imaging, where autofluorescence can be generated from background animal tissues by excitation light, bioluminescence imaging can produce more specific and stronger signals with a longer exposure time.
Short-Term Labeling
In studies that observe HSC homing to the bone marrow niche after transplantation, identifying the location of donor cells and distinguishing them from endogenous cells are important. Given that the HSC homing process can be short (ie, hours to days), HSCs have been frequently labeled ex vivo with a fluorescent probe before they are transplanted into a recipient animal.14,28–30 A series of fluorescent dyes can be used for cell tracing, which includes carbocyanine dyes (such as dioctadecyl tetra-methylindodicarbocyanine [DiD], or dioctadecyl tetra-methylindotricarbocyanine [DiR]), PKH26, Hoechst 33342, 4′,6-diamidino-2-phenylindole (DAPI), and BrdU.
Lipophilic membrane dyes, such PKH26 and DiD, have been used to label HSCs harvested from the donor bone marrow without marked alteration of HSC functions.14,28–30 The fluorescence of carbocyanine dyes is greatly enhanced after incorporation into the cell membrane than in free forms. Compared to dioctadecyl tetramethylindocarbocyanine (DiI) and dioctadecyloxacarbocyanine (DiO) in the same family, DiD can be excited by a lower-power heliumneon laser or a red diode laser in the 630 to 650 nm range. Its 665 nm emission also propagates well through animal tissues and blood. DiR is another dye that might be better suited for in vivo imaging or tracing because the autofluorescence background in tissues is much lower at the DiR emission wavelength of 780 nm. These dyes can be applied to label stem cells directly or to label other cell types for multicolor imaging. The drawbacks of membrane dyes include certain cell toxicity 31 and short windows for imaging owing to cell division-mediated signal reduction.28,31
DNA-binding dyes such as Hoechst 33342 and DAPI have a high labeling efficiency and can be used as tracking dyes for short-term cell migration. 32 Both dyes can be excited by ultraviolet (UV) light, and their blue emission (at ≈461 nm) is convenient for co-imaging with other fluorescent stains. Labeling by a thymidine analogue, BrdU, has also been used to label dividing cells, including hematopoietic stem and progenitor cells. 17 One of the major disadvantages of the DNA-binding dyes is that they can be taken up by neighboring cells after being released by dead cells, therefore resulting in false-positive data.18,19 It has also been reported that BrdU labeling may alter stem cell proliferation and differentiation 20 and BrdU retention has poor sensitivity and specificity as a stem cell marker. 21
More recently, the use of nanoparticles such as quantum dots has attracted much attention as a new generation of labeling reagents. 33 Quantum dots have a higher molar extinction coefficiency than the aforementioned fluorescent dyes; therefore, they can produce brighter signals. The improved photostability of quantum dots allows acquisition of images for a prolonged period of time. Given that their optical properties correlate with their sizes, it is possible to choose quantum dots (7–10 nm) that emit at near-infrared wavelength to take advantage of the improved tissue penetration and lower autofluorescence in the tissue at these wavelengths, although the low wavelength of excitation still limits the depth of tissue penetration. 34 All of these properties make them attractive choices for in vivo imaging.
Noninvasive Short-Term Labeling
Because physical labeling is convenient and can be completed within hours, most short-term noninvasive imaging has relied on physical labeling, such as superparamagnetic iron oxide (SPIO) particles for MRI 24 and radionuclide labeling for single-photon emission computed tomography (SPECT) or positron emission tomography (PET). 35 Quantum dots can also be designed and manufactured to contain both optical and magnetic properties suitable for noninvasive imaging. MRI can be used to locate the transplanted stem cells with a high spatial resolution (25–100 μm), and SPECT and PET have high sensitivity. Therefore, such physical labeling methods are suitable for short-term imaging studies such as monitoring initial trafficking of stem cells posttransplantation. One potential drawback of physical labeling is cellular toxicity as it has been reported that certain cell functions can be altered on intake of nanoparticles.36–38 Long-term survival, differentiation, or proliferation cannot be imaged by physical labeling because these labels would be diluted during cell division and radioisotopes would decay over time.
Long-Term Labeling
The most popular labeling of stem cells and their derivatives for long-term cell-fate studies is through expression of transgenes encoding fluorescent proteins. Given that the transgene expression cassette can be integrated in the genome through lentiviral or retroviral transduction, it will not be lost through cell divisions. Nonintegrated gene expression (through adenoviral vectors, for example) may be sufficient for certain in vivo localization studies 39 but is not suitable for long-term tracking. The expression can be constitutive by using a housekeeping gene promoter or can be tissue specific by enhancer elements that are selectively activated in certain types of cells. For example, stem cells isolated from green fluorescent protein (GFP) transgenic mice can be transplanted into recipients that do not carry this transgene, allowing monitoring of GFP donor signals for long-term survival, proliferation, and distribution studies.40,41 Alternatively, transgenic animals can be created with fluorescent protein coding genes under the control of a promoter specific to the cell type of interest. An example of such a strategy is the CD41:GFP transgenic mouse or zebrafish, in which GFP gene expression correlates with expression of CD41, one of the specific marker genes for the earliest HSCs during blood development. 42 Early hematopoietic development can be monitored by the emergence of cells with a GFP signal in such animals. 42
One disadvantage of this approach is that well-defined tissue-specific promoters are not available for every cell type. Another disadvantage of using this approach to study gene expression is that some fluorescent proteins, including GFP, are relatively stable inside cells even a few days after the expression of the reporter gene has been shut down. Therefore, imaging results may not accurately reflect the real gene expression. On the other hand, this fluorescent protein retention phenomenon has been taken advantage of to visualize skin stem cells tagged with histone H2B-GFP in transgenic mice. 43 After transgene expression was switched off by tetracycline administration, the remaining GFP would be diluted as cells divide. Only quiescent stem cells undergoing slow or no cell cycling would retain their GFP signal, allowing localization of these stem cells in the bulge area of hair follicles. 43
Another commonly used histochemical reporter is the bacterial enzyme β-galactosidase (LacZ). LacZ-expressing cells can be easily located using the substrate X-gal, which turns blue when it is catalyzed by β-galactosidase.15,44 The live imaging of transgenic cells expressing the bacterial LacZ reporter gene has been hindered by the presence of mammalian forms of β-galactosidases in many adult cell types and the problematic nature of fluorogenic LacZ substrates getting into live cells.
In stem cell transplantation studies, the Y chromosome has also been used for cell tracing because the location of the implanted cells in recipients can be detected by fluorescence in situ hybridization (FISH) analysis after cells derived from male donors were transplanted into the female recipients.14,45–48 Compared to gene expression of GFP, the Y chromosome tracing technique is a very straightforward process with a higher labeling efficiency. In recent years, it has been widely used in stem cell transplantations for liver, cardiac, and intestinal disease as well as skin injury to identify the implanted stem cells.14,45–48 However, it is limited only to sex-mismatched studies that often require transplantation of male (XY) donor cells to female (XX) recipients.
Noninvasive Long-Term Labeling
The transgene approach can also be used for long-term noninvasive imaging. The most commonly used reporter gene for bioluminescence imaging is the firefly luciferase gene (fLuc). Photons can be generated in Luc-expressing cells after systemic administration of luciferase substrate
Genetic labeling of the cells with a transgene is required for these approaches. It is technically feasible to label pluripotent stem cells with a high efficiency. 52 Immortalized neuroprogenitor cells have also been stably transfected with a luciferase gene, and these cells were used to study the migratory capacities of neuroprogenitors toward brain tumors after implantation. 53 It remains challenging to efficiently label primary adult stem cells (eg, HSCs), which are rare and hard to expand in tissue culture, although significant advances have been made in the past few years.54,55 Moreover, although the bioluminescence or radioisotope-based imaging is excellent technology for providing general anatomic locations of transplanted cells over time, the spatial resolution is relatively low, not suitable for a cell-cell interaction study.
Recent Advances in Stem Cell Research Using Imaging Technology
Elucidating the Developmental Origins of HSCs
The developmental origins of HSCs have been debated, especially the relationship of HSCs to blood vessel-forming endothelial cells. Both types of cells arise from mesoderm (one of the three embryonic germ layers) during embryonic development, and it has been proposed that they are closely related and can be traced back to a common precursor, termed the hemangioblast. 56 Sectioning and staining of fixed embryonic tissues as well as cell isolation and functional transplantation experiments have revealed a close temporal and spatial relationship between these two cell types in the ventral wall of the dorsal aorta around E10.5 in mice and 7 weeks in humans.57,58 Clusters of HSCs have been observed hanging outside the endothelial walls. Questions remain regarding at which stage these two lineages diverge. More recently, a series of exciting studies using lineage tracing have suggested that HSCs go through an endothelial stage; thus, the term hemogenic endothelium was proposed as the direct precursor of HSCs. 44
To study this process in more detail, several groups used in vitro imaging techniques to monitor the budding of HSCs from the endothelium. By imaging the in vitro cultured fetal liver kinase 1+ (Flk1+) mesodermal cells that were generated from day 4 differentiation of mouse embryonic stem cells, Lancrin and colleagues analyzed the sequential cellular events leading to the generation of HSCs. 59 This time-lapse photography study revealed that a population representing hemangioblasts (Flk-1+) gave rise to cells expressing various endothelial markers at about 48 hours after culture, and this event was followed by the emergence of HSCs from the endothelium-containing clusters. 59 In a separate study, Eilken and colleagues also analyzed mouse embryonic stem cell differentiation on OP9 stromal cells. 60 By continuous single-cell imaging, they tracked the fates of thousands of cells during differentiation. Living endothelial and hematopoietic cells were identified by simultaneous detection of morphology and multiple molecular and functional markers. It was observed that some adherent endothelial cells directly give rise to nonadherent HSCs. 60 These in vitro imaging studies convincingly demonstrated that hemogenic endothelial cells can be generated from mouse embryonic stem cells. However, evidence for natural occurrence of this process during embryonic development has been lacking.
More recently, several groups took on this challenge by directly imaging the aortic endothelium region in live zebrafish embryos and live sections of mouse embryos.42,61,62 These groups took advantage of time-lapse confocal and two-photon fluorescence microscopy technologies to visualize cells residing deeper within tissues over time. Taking advantage of the relative transparency of zebrafish embryos and transgenic zebrafish that would express enhanced GFP (eGFP) in definitive HSCs and mCherry (an improved monomeric red fluorescent protein) in endothelial cells (cmyb:eGFP; kdrl:mCherry), Bertrand and colleagues observed that cMyb:eGFP+ cells arose directly from Kdrl:mCherry+ cells, specifically along the ventral aspect of the dorsal aorta. 62 Using a similar strategy and a kdrl: GFP transgenic zebrafish, Kissa and Herbomel recorded the detailed cell transition, termed endothelial hematopoietic transition, in which certain endothelial cells from the aortic floor undergo lasting contraction, cell bending, rounding up, and eventually leaving the vascular floor without compromising the vessel's integrity. 61 By monitoring a different transgenic zebrafish, which was labeled by Lmo2:DsRed and CD41:GFP, they confirmed that the budding of cells from the aortic floor is accompanied by the onset of CD41 expression, an early hematopoietic marker, suggesting that the cells are going through endothelial to hematopoietic fate transition. Given that mouse embryos are significantly thicker and less transparent than zebrafish, currently, there is no available technology that allows the direct imaging of inside regions such as the dorsal aorta at a single-cell level. Boisset and colleagues developed a new experimental approach of cutting mouse embryos (E9 to E11) into thick transverse sections to overcome this technical problem. 42 Using this strategy, the architecture and organization of the aorta and surrounding tissues were conserved, allowing visualization of live cells in the dynamic and physiologic context of the aorta. By using different hematopoietic reporter transgenic mice (such as Ly-6A-GFP and CD41-YFP), as well as antibody staining of the embryos with various antibodies marking endothelial and hematopoietic cells, the authors observed the dynamic de novo emergence of phenotypically defined HSCs directly from ventral aortic hemogenic endothelial cells. These imaging studies in zebrafish and mouse embryos provide firsthand evidence that hematopoietic development can go through the hemogenic endothelial stage. It also provides systems to further identify critical molecular regulators of this process; ultimately, the knowledge will aid our efforts to derive patient-specific HSCs from pluripotent stem cells for therapeutic and disease modeling purposes.12,63
Detection of HSC Niche in Adult Bone Marrow
After its generation from the aorta-gonad-mesonephros region, HSCs migrate through the fetal liver and eventually reside in bone marrow in the adult. Understanding how these cells are regulated to undergo self-renewal, expansion, or differentiation is among the most important tasks in stem cell biology. However, owing to the complexity of the bone marrow environment and the rarity of HSCs, it has not been clear where these cells physically reside and what cell types are in direct contact with them. It has been difficult to directly visualize the live bone marrow environment through conventional microscopy. Lo Celso and colleagues recently tackled this problem by using a combination of high-resolution confocal microscopy and two-photon video imaging of individual HSCs in the calvarium bone marrow (up to 150 μm in depth) of living mice over time. 30 To simultaneously detect multiple cell types in bone marrow, several labeling strategies were used: (1) osteoblasts were labeled by using osteoblast-restricted collagen 1α promoter (Col2.3-GFP) reporter mice; (2) quantum dots were injected immediately before imaging to label vasculature; (3) bone structure was detected by second harmonic generation microscopy; and (4) HSCs were labeled with DiD or DiI. This multichannel two-photon approach for live imaging through the thin bone provides higher-resolution single-cell images within the marrow cavities. It is therefore possible to image relatively few transplanted stem cells and their precise location with respect to the niche. Together with another study, performed ex vivo, in which a confocal laser scanning microscope was used to image GFP-labeled mouse HSCs in real time within bone marrow, 40 these reports demonstrated the important role of the endosteal niche for HSCs, at least in the setting of BMT. The relationship between HSCs and the bone marrow niche components (ie, osteoblasts or vasculature) is still difficult to study by imaging owing to the constraints of the current in vivo imaging technology. In addition, the imaging depth in these studies is limited to about 150 μm below the bone surface. Thus, it is difficult to image the long bones, such as the femur and tibia. Moreover, in most cases, only one or two follow-up imaging sessions are possible owing to surgery-related scar formation. 64
Noninvasive Imaging of Human ES Cell and Derivatives after Transplantation
Directed differentiation of pluripotent stem cells to functional tissue lineages provides alternatives to adult stem cells as sources for transplantation. It is therefore important to track the migration, engraftment, proliferation, and differentiation of these cells in vivo. Because pluripotent stem cells have the ability to form a teratoma, it is also essential to develop a sensitive technology to monitor the tumor formation process.
Given that transient labeling methods do not sustain the signal long enough through extensive cell divisions after transplantation, permanent cell marking by the expression of reporter genes is preferred. For this purpose, lentiviral vectors that are most resistant to silencing of a transgene are used to constitutively express a reporter gene, followed by noninvasive live imaging to track the cell implantation and tumor formation after transplantation of lentiviral labeled human cells in immunodeficient mice.49–51 hES cells can be labeled by a reporter transgene encoding either fLuc for bioluminescence imaging or the HSV1-tk for radiopharmaceutical-based imaging. The implanted hES cells expressing the fLuc gene can be repetitively monitored after systemic administration of
However, it is important to note two potential caveats of this approach. First, it is reported that

Detection of engrafted cells in live mice after transplantation of human embryonic stem cells labeled with a lentiviral vector expressing the firefly luciferase gene. A, A diagram of an integrating lentiviral vector coexpressing the firefly luciferase gene (fLuc) and puromycin resistance gene (PuroR) under the control of human ubiquitin C (Ubc) gene promoter/enhancer. B, In vivo imaging of two immunodeficient mice (left) and three immunocompetent mice (right) 7 days after implantation of human embryonic stem cells that have been transduced with the luciferase-expressing lentivirus.
The basic platform of the lentivirus-mediated reporter expression technology used in these studies also allows tissue-specific imaging by controlling fLuc gene expression by a tissue-specific promoter. To extend live imaging technology to relatively large animals or patients, we have also used clinically translatable in vivo imaging technologies, such as SPECT, to detect hES cells expressing HSV1-tk after transplantation into immunodeficient (SCID) mice. 50 Systemic administration of HSV1-tk substrate 125I FIAU allowed sensitive and quantitative imaging of the implanted hES cells. 50 Similar strategies have also been used to image the in vivo behavior of mouse emybronic stem cell derivatives16,66 and endothelial cells derived from hES cells.49,51
Live Imaging of Dynamic Marker Expression during Human iPS Cell Generation
Although the first human iPS cell lines were derived only less than 3 years ago, this technology has already made a significant impact on stem cell research and regenerative medicine.9,10 It is anticipated that patient-specific iPS cells will serve as ideal sources for cell replacement therapy and for disease modeling. However, this technology is still in its early stage, and many questions still need to be addressed before iPS cells can be widely used. One of the key issues is to identify bona fide iPS cells from other transformed cells during the reprogramming process. Many cell surface markers and pluripotency-related genes have been used to characterize undifferentiated hES cells or iPS cells, including alkaline phosphatase (APase), SSEA4, SSEA3, TRA-1-60, TRA-1-81, OCT4, SOX2, NANOG, GDF3, and REX1.9–13 Unlike the mouse system, where various transgenic strains can be created and reporter genes have been helpful in identifying fully reprogrammed cells, human iPS cell identification has to rely on cell morphology and surface marker expression. To define molecular markers that can reliably identify human iPS colonies among a large number of transformed cells at early stages of iPS reprogramming, Chan and colleagues traced and analyzed reprogramming human fibroblast cultures with multicolor live immunofluorescence imaging by automated fluorescence microcopy scanning. 67 Serial live cell imaging of emerging colonies by staining in situ with antibodies revealed that TRA-1-60 detection and expression of DNMT3B and REX1 can be used to distinguish fully reprogrammed states. In comparison, APase and SSEA4 expression arose earlier but was insufficient to signify the full reprogramming leading to bona fide iPS cells. 67 Our reprogramming studies independently confirmed that the combination of TRA-1-60 and morphologic criteria (hES-like morphology) is a more reliable indicator than other pluripotency-related markers, such as APase and SSEA4, not only for fibroblasts but also for blood cells and cell types from nonmesodermal origins.13,68 The serial live immunofluorescence scanning and imaging technology used in this study are also likely to be useful for studying cellular events during in vitro iPS differentiation to defined lineages.
Future Challenges
State-of-the-art imaging technologies have been instrumental in identifying the developmental origin of HSCs and the bone marrow microenvironment for adult HSCs. They are also essential for investigating the in vivo behavior of transplanted cells, including adult stem cells and embryonic stem/iPS cell derivatives. For developing a safe and effective cell replacement therapy, novel molecular imaging techniques to longitudinally and precisely monitor stem cell fate (ie, distribution, localization, migration, proliferation, and differentiation) in living subjects are highly desired. As discussed, a reporter gene expression approach is one of the most reliable methods to achieve long-term cell labeling. Tissue-specific expression of reporters would tremendously benefit the studies of stem cell fate determination. However, there are currently limited numbers of true tissue-specific promoter/enhancer elements that can be practically used for controlling transgene expression. An alternative way is to knock in the reporter gene to a known tissue-specific gene locus by homologous recombination (HR)-mediated gene targeting. This is also a highly desired technology, and we have shown that it is possible to improve HR efficiency in hES cells by a pair of zinc-finger nucleases that make double-strand breaks at a specific DNA sequence. 69 Accumulating knowledge regarding promoter structures and gene targeting will no doubt aid our efforts, although indirectly, in enhancing stem cell imaging.
Nanoparticles such as SPIO and quantum dots have shown promising potential for molecular imaging. The fast-developing quantum dot technology will likely create more and better imaging modalities in the near future. It will also be important to identify strategies to retain nanoparticle-mediated cell labeling to allow longer-term imaging. At the same time, little has been done to analyze the effects of nanoparticle labeling on stem cell function (eg, self-renewal and differentiation). Comprehensive studies using various types of stem cells are required to demonstrate the safety of this technology.
The development of multiphoton laser scanning microscopy significantly enhanced our ability to image cells in live tissues. It will continue to play an increasingly important role in stem cell biology. However, the current imaging depth is still insufficient, and most of the tissues (eg, femur) cannot be directly imaged without being surgically removed from the body. Although bioluminescence imaging has a much better penetration capability, it lacks a single-cell resolution that is important for many studies involving cell-cell interactions. Thus, developing a novel, high-resolution, noninvasive imaging modality would be one of the most challenging but also highly rewarding tasks.
Footnotes
Acknowledgments
We thank Dr. Saul Sharkis and Mr. Michael Collector for critical reading of the manuscript and members of the Jang and Cheng laboratories for sharing their unpublished results. We thank Dr. Liuhong Cai for her assistance in the preparation of the manuscript. We regret that we are unable to cite other relevant studies because of space constraints.
Financial disclosure of authors: Dr. Ye is supported by a National Institutes of Health postdoctoral fellowship (T32 HL007525). Dr. Jang's group is supported by grants from the Maryland Stem Cell Research Fund and the National Institute of Diabetes and Digestive and Kidney Diseases, and Dr. Cheng is supported by grants from the National Heart, Lung and Blood Institute.
Financial disclosure of reviewers: None reported.