
Abstract
Nuclear factor κB (NF-κB) is a transcription factor that plays a major role in many human disorders, including immune diseases and cancer. We designed a reporter system based on NF-κB responsive promoter elements driving expression of the secreted Gaussia princeps luciferase (Gluc). We show that this bioluminescent reporter is a highly sensitive tool for noninvasive monitoring of the kinetics of NF-κB activation and inhibition over time, both in conditioned medium of cultured cells and in the blood and urine of animals. NF-κB activation was successfully monitored in real time in endothelial cells in response to tumor angiogenic signaling, as well as in monocytes in response to inflammation. Further, we demonstrated dual blood monitoring of both NF-κB activation during tumor development as correlated to tumor formation using the NF-κB Gluc reporter, as well as the secreted alkaline phosphatase reporter. This NF-κB reporter system provides a powerful tool for monitoring NF-κB activity in real time in vitro and in vivo.
OVER TWO DECADES AGO, Sen and Baltimore described a nuclear factor that binds to deoxyribonucleic acid (DNA) sequences in the promoter and enhancer regions of the mouse immunoglobulin gene. 1 This transcription activation mechanism, originally thought to occur exclusively in B cells, soon turned out to be a universal transcription process with important roles in many physiologic disorders. This transcription factor is referred to as nuclear factor κB (NF-κB). NF-κB is a heterodimeric protein composed of different combinations of five members of the Rel family of transcription factors, including p65 (RelA), RelB, c-Rel, p50, and p52, containing a common Rel homology domain (RHD) within their N-terminal region. 2 The RHD is responsible for DNA binding as well as homo- and heterodimerization. Only p65, RelB, and c-Rel contain a transcription activation domain allowing the induction of gene expression. 3 The NF-κB protein complex is usually present in the cytoplasm as homo- or heterodimers bound to the IκB family (mainly IκBα). Binding of IκB prevents NF-κB translocation from the cytoplasm into the nucleus and subsequent binding of NF-κB to the DNA. 4 A wide variety of signals, such as proinflammatory cytokines, growth factors, hormones, oxidative stress, viral infection, and DNA-damaging agents, 5 can cause the activation of IκB kinase (IKK), which, in turn, phosphorylates the IκB, causing the release of the NF-κB dimers and their nuclear translocation. In the nucleus, the NF-κB dimers bind to a κB site in the promoter or enhancer region of target genes, thereby controlling gene expression. Activated NF-κB can induce the transcription of many genes, such as cytokines, growth factors, adhesion molecules, and mitochondrial antiapoptotic genes. 2
Although the crucial role of NF-κB in the immune response is well established, 6 cumulative evidence has shown that it is a key mediator in inflammation as well as in tumor development, progression, and neovascularization. 7 Constitutive NF-κB activation has already been demonstrated in many cancer types.8–10 Activation of this signaling pathway can lead to the transcription of many antiapoptotic genes and the inhibition of apoptosis, causing drug resistance. 11 Further, NF-κB has been shown to play an important role in tumor angiogenesis and invasiveness. 12
Bioluminescence imaging has emerged as a powerful tool in biomedical research for monitoring of transgene expression, viral vector infection, tumor growth, and metastasis, as well as inflammation and gene therapy. 13 Recently, we established a novel assay for ex vivo bioluminescence measurement of in vivo processes. 14 By cloning the naturally secreted Gaussia princeps luciferase (Gluc) under the control of the constitutively active cytomegalovirus (CMV) promoter, we were able to monitor different biologic processes, such as tumor growth and response to therapy, as well as tracking of circulating cells by assessing the Gluc activity in a few microliters of blood, urine, or serum in mice. 14 We constructed an NF-κB reporter system comprising five tandem repeats of the NF-κB transcription responsive elements (TREs; 12 bp each) and a TATA box driving the expression of the secreted Gluc. We showed that Gluc expression was induced up to 500-fold in response to NF-κB activation in response to tumor necrosis factor α (TNF-α) and was inhibited by 5-fold in response to sulfasalazine (SSZ) in a dose- and time-dependent manner in vitro. Further, we successfully monitored NF-κB activation in real time in response to different stimuli, including tumor angiogenesis, as well as in circulating monocytes on induction of inflammatory responses. In addition, using two secreted enzyme reporters requiring different substrates (Gluc and the secreted alkaline phosphatase [SEAP]), we established a dual blood assay system allowing the monitoring of NF-κB activation during tumor development as correlated with cancer cell proliferation and tumor growth.
Materials and Methods
Vector Construction
Five tandem repeats of NF-κB TREs (TGGGGACTTTCCGC)15,16 were designed and annealed using the complementary oligonucleotide sequences 5′-GATCTTGGGGACTTTCCGCTGGGGACTTTC CGCTGGGGACTTTCCGCTGGGGACTTTCCGCT GGGGACTTTCCGCA-3′ and 5′-CTAGTGCGGAAAGTCC CCAGCGGAAAGTCCCCAGCGGAAAGTCCCCAGC GGAAAGTCCCCAGCGGAAAGTCCCCAA-3′. Additional NF-κB reporters with multiple tandem repeats of 5, 10, and 15 NF-κB TREs were constructed with 0, 4, 8, 12, or 30 random basepairs acting as spacers separating each of the tandem copies (Figure 1A). All of these promoter elements were designed to generate sticky ends compatible with BglII and NheI restriction sites at the 5′ and 3′ ends, respectively The humanized Gaussia princeps luciferase complementary DNA (cDNA) (Nanolight, Pinetop, AZ) 17 was amplified by polymerase chain reaction, adding a TATA box from the human immunodeficiency virus (HIV) type 1 subtype E promoter (ATATA) 30 bp upstream of the Gluc cDNA. The TATA-Gluc fragment was purified from a 1% agarose gel and ligated into the mammalian expression plasmid pHGCx 18 (from Dr. Yoshimura Saeki, Massachusetts General Hospital, Boston, MA) by replacing the CMV promoter with NF-κB elements generating pHGNF-Gluc. Similarly, the CMV promoter in pHGCX was replaced with the SV40 promoter (amplified from pSEAP2-control plasmid, Clontech, Palo Alto, CA) generating pHGSV40-Gluc. The NF-Gluc expression cassette was subsequently subcloned into CSCW, a self-inactivating lentivirus vector, 19 at the BamHI and XhoI sites producing pCSNF-Gluc. A DNA fragment spanning two copies of the 1.2 kb chicken β-globin (HS4) insulator elements, amplified from the pJC13-1 plasmid 20 (a kind gift from Dr. Adam West, National Institutes of Health, Bethesda, MD), was inserted into the U3 region of the pCSNF-Gluc plasmid using the PmeI and KpnI restriction sites creating pCSHSNF-Gluc (designated lenti-NF-Gluc throughout the text). Using the pCSCW-IG self-inactivating vector, 19 we cloned the mCherry fluorescent protein (kindly provided by Dr. Roger Tsien, University of California-San Diego, CA) cDNAs under the control of the CMV promoter, thereby generating LV-mCherry. SEAP and mCherry separated by an internal ribosome entry site were cloned in a similar vector by amplifying the SEAP from the pSEAP2-basic vector (Clontech). 14 The integrity of all constructs was verified by sequencing. All lentivirus vectors were produced and titered as transducing units (TU)/mL as previously described. 19
Cell Culture and Reagents
293T human kidney fibroblast cells (from Dr. Maria Calos, Stanford University, Stanford, CA), U87 human glioma (American Type Culture Collection [ATCC], Manassas, VA; U87 MG), Gli36 human glioma (from Dr. Anthony Capanogni, University of California-Los Angeles, Los Angeles, CA), 21 A549 human lung carcinoma (ATCC), and U937 monocytic/histiocytic lymphoma (ATCC) cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Sigma, St. Louis, MO), 100 U/mL penicillin, and 0.1 mg/mL streptomycin (Sigma). HEI193 human schwannoma cells 22 were maintained in DMEM supplemented with 10% FBS, 2 μM forskolin (Calbiochem Corp., LaJolla, CA), 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 50 mg/mL gentamicin (Invitrogen, Carlsbad, CA). U87-CFP cells were produced by stably transducing U87 cells with a CMV-controlled Cerulean expression cassette using a lentivirus vector. 23 Human brain microvascular endothelial cells (HBMVECs; Cell Systems ACBRI-376, Kirkland, WA) were cultured in endothelial growth medium (EGM) (Cambrex, San Diego, CA) for no more than 10 passages.
HBMVEC-mCherry cells were produced by stably transducing HBMVECs (passage 5) with lentivirus vector expressing mCherry. After three passages, the cells were discarded. All cells were grown at 37°C in a 5% CO2 humidified incubator. SSZ, parthenolide (PTL), etoposide, and doxorubicin were purchased from Sigma. Recombinant human TNF-α was obtained from R&D Systems (Minneapolis, MN). Ionizing radiation (IR) treatments were performed at 3 to 10 Gy using a 137Cs source.

The nuclear factor κB (NF-κB)-Gluc reporter system. A, Schematic overview of the different NF-Gluc expression cassettes with the corresponding Gluc induction levels. 293T cells were transfected with plasmids carrying the expression cassette of Gluc under the control of different NF-κB responsive elements. Cells were either not treated or irradiated with 5 Gy of ionizing radiation, and 24 hours later, Gluc activity was measured in an aliquot of conditioned medium. B, 293T cells were transduced with lentivirus vector encoding Gluc under NFκB-0 transcription responsive elements with or without a TATA box. Cells were treated with tumor necrosis factor α (10 ng/mL), and Gluc activity was measured in an aliquot of the conditioned medium 24 hours later. C, 293T cells were transduced with lentivirus vector expressing 5NFκB-0-Gluc with or without HS4 insulator elements. Forty-eight hours later, Gluc was measured in an aliquot of the conditioned medium. Data in B and C are presented as the average fold increase in Gluc activity ± SD (n = 4). ** p ≤ .01, Student t-test.
Transfection and Lentivirus Transduction
293T human fibroblast cells were plated in a six-well plate (1.5 × 104 cells/well) and transfected with plasmids expressing Gluc under either NF-κB, CMV, or SV40 promoters using Lipofectamine 2000 (Invitrogen) according to the manufacturer's guidelines. To achieve stable gene expression in most cells (> 90%), different cell types were transduced with lentivirus vectors at 100 TU/cell.
In Vitro Gaussia princeps Luciferase Activity
For Gluc assays, 15 μL aliquots of the cell-free conditioned medium were collected at different time points. Gluc activity was assayed by adding 20 μM coelenterazine, the Gluc substrate (Nanolight), to the supernatant and measuring photon counts in a 96-well plate luminometer (Dynex, Richfield, MN) over 10 seconds.
In Vitro Angiogenesis Assay
Low-passage HBMVEC-mCherry cells transduced with lentivirus vector to express NF-Gluc (passage < 7) were cultured on Matrigel (Beckton Dickinson, San Jose, CA) in EBM basal medium (Cambrex) in the presence or absence of U87-CFP cells or EGM cocktail (Cambrex). Twenty-four hours later, aliquots of conditioned medium were analyzed for Gluc activity and the cultures were analyzed by a combination of light and fluorescence microscopy.
In Vivo Experiments
All animal experiments were approved by the Massachusetts General Hospital Subcommittee on Research Animal Care. One million HEI193, 293T, or Gli36 expressing NF-Gluc and/or SEAP cells were injected subcutaneously into nude mice. TNF-α (16 μg/kg body weight) and SSZ (15 mg/kg body weight) were injected intravenously unless otherwise specified. For circulating tumor cells, 0.5 million cells expressing NF-Gluc and SEAP were injected intraocularly.
Monocyte Model
U937 human leukemic monocyte lymphoma cells were transduced with lenti-NF-Gluc at 100 TU/cell. Cells were plated in 96-well plates and treated with different concentrations of human TNF-α (R&D Systems). At different time points, Gluc activity was assayed in the medium as above. For the in vivo studies, 1 million U937 cells expressing NF-Gluc were injected intraperitoneally into eight mice, and 1 hour later, four mice were injected with phosphate-buffered saline (PBS) (control) and the other four mice with 2 μg of TNF-α (in 100 μL) via the same route. Before PBS or TNF-α injection and at different time points after injection, 5 μL blood was withdrawn and assayed for Gluc activity (see below).
Gluc/SEAP Blood Assay
For the Gluc assay, 5 μL of blood was collected by making a small nick in the mouse tail and was mixed immediately with 1 μL 20 mM ethylenediaminetetraacetic acid (EDTA). Gluc activity was measured using the luminometer after injecting 100 μL 100 μM coelenterazine and acquiring photon counts over 10 seconds. The SEAP chemiluminescence activity was measured in 5 μL serum using the Great EscAPe SEAP assay kit (Clontech) as per the manufacturer's instructions using the luminometer.
In Vivo Bioluminescence Imaging
Mice were anesthetized by intraperitoneal injections of ketamine (100 mg/kg) and xylazine (5 mg/kg) and then received an intraocular injection of coelenterazine (4 mg/kg body weight diluted in PBS). Photon counts were acquired for 5 minutes using a cooled charge-coupled device (CCD) camera system. White-light surface images were also taken before each Gluc imaging session, providing an anatomic view of the animal. Image processing and signal intensity quantification and analysis were performed using CMIR-Image, software developed by the Center for Molecular Imaging Research at Massachusetts General Hospital using image display and an analysis suite developed by IDL (Research Systems Inc., Boulder, CO). Images were represented in a pseudocolor photon count manner, superimposed on a grayscale anatomic white-light image, exposing the bioluminescence intensity and the animal anatomy. Bioluminescence signals were defined by an automatic intensity highlight procedure after subtracting background noise. The sum of the photon counts was calculated as a measure of Gluc reporter activity.
Results
Construction of the NF-Gluc Reporter
Initially, we designed and compared various NF-κB responsive promoters by cloning multiple tandem repeats of the NF-κB TRE with a different linker between each of the repeats (to reduce potential steric hindrance) to drive the expression of the Gluc reporter (see Figure 1A). 293T cells were transfected with each of these reporters, and 24 hours later, NF-κB was activated either by IR 24 or TNF-α. 25 Gluc induction levels in response to NF-κB activation were assessed in aliquots of conditioned medium 24 hours posttreatment. A significant increase in Gluc expression was detected after stimulation of these cells with IR for all six different constructs, with 5NFκB-0 and 2 * 5NFκB giving sixfold induction (see Figure 1A). Adding more than five tandem repeats of the TRE or different linkers between each repeat did not enhance the Gluc induction level; therefore, we decided to proceed with the construct containing five TREs of NF-κB with no spacers (5NFκB-0) in all subsequent studies. To enhance the transcriptional activity of the reporter, a TATA box was inserted downstream of the NF-κB TRE. As expected, a > 28-fold increase in Gluc induction was observed in response to NF-κB activation on TNF-α treatment compared with the reporter without the TATA box (Figure 1B). When we packaged the NF-Gluc into a lentivirus vector and transduced 293T cells, we noticed a high basal level of Gluc expression compared with transient transfection. To reduce any possible NF-κB-independent transactivation of Gluc expression, we inserted chicken β-globin insulators 20 in the U3 region of the lentivirus vector, thereby flanking the Gluc expression cassette on expression. This vector was designated NF-Gluc. The addition of the insulators showed a much tighter system with a significant reduction in the Gluc basal level in the absence of NF-κB activating stimuli (Figure 1C; ** p ≤ .01).
In Vitro Monitoring of NF-κB Activation and Inhibition
To confirm that the increase in Gluc expression is due to NF-κB activation, 293T cells were transfected with plasmids expressing Gluc under either NF-κB TRE, SV40, or CMV promoters and treated with TNF-α (a renowned inducer of NF-κB activity 25 ) or PBS as a control. Twenty-four hours later, 20 μL aliquots of conditioned medium were transferred into a 96-well plate and assayed for Gluc activity using the luminometer (Figure 2A). The plate was also imaged using the CCD camera (Figure 2B). As expected, cells expressing Gluc under the control of NF-κB TREs showed a > 100-fold increase in Gluc expression in response to TNF-α, indicating NF-κB-mediated transcriptional activation of the NF-Gluc reporter (see Figure 2, A and B). On the other hand, the SV40 promoter did not show any increase in Gluc activity in response to TNF-α, whereas, surprisingly, the CMV promoter showed around a twofold increase in Gluc expression in response to TNF-α. These results support previous reports that the CMV promoter can be induced by different drugs and that TNF-α can further stimulate the activation of the CMV promoter via induction of NF-κB.26,27
To test whether the NF-Gluc reporter system could be used to monitor NF-κB activation in response to different stimuli, 293T human embryonic kidney cells were transduced with a lentivirus vector expressing NF-Gluc (lenti-NF-Gluc) to stably express Gluc under the control of NF-κB. These cells were treated with different drugs previously described to induce NF-κB activation, the inflammatory cytokine TNF-α, 25 the chemotherapeutic drugs etoposide and doxorubicin, 28 and the radiomimetic drug bleomycin sulfate. 29 Alternatively, cells received a single dose of 5 Gy of IR. 24 The Gluc expression was assayed 24 hours posttreatment. The increase in Gluc expression as a measure of NF-κB activation varied among the different treatments, with TNF-α resulting in a > 500-fold induction (Figure 2C). To investigate the activation of this reporter in different cell types, we transduced different human tumor cell lines, Gli36 glioma cells, HEI193 schwannoma cells, and A549 lung adenocarcinoma cells, as well as 293T human fibroblast cells, with lenti-NF-Gluc. These cells have > 95% transduction efficiency with lentivirus vector under the conditions used.30,31 These cells were treated with TNF-α, and the Gluc activity was measured in an aliquot of conditioned medium 24 hours later. As expected, HEI193 human schwannoma cells (a benign tumor32–35) and 293T fibroblast cells showed the lowest basal levels of NF-κB activity (around 500 RLU) and consequently the highest Gluc induction levels (> 100-fold; Figure 2D). On the other hand, Gli36 human glioma and A549 human lung carcinoma cells (both malignant and invasive tumor types36,37) presented with high basal NF-κB activity (around 120,000 RLU; data not shown) and a moderate additional increase in Gluc expression (three- to fivefold) on TNF-α treatment (see Figure 2D).
To determine the usefulness of the NF-Gluc reporter in monitoring NF-κB activation in a dose- and time-dependent manner and in real time, 293T cells stably expressing NF-Gluc were treated with different concentrations of TNF-α. The Gluc signal increased on increasing the TNF-α dose (Figure 2E). Notably, even the lowest concentration (75 pg/mL) resulted in a > 50-fold induction. When conditioned medium was collected at different time points and assayed for Gluc activity, an increase was observed as early as 2 hours after TNF-α treatment, reaching a maximum at 48 hours (Figure 2F).
SSZ, an antiinflammatory and immunosuppressive agent, inhibits IKKα and IKKβ, thereby blocking NF-κB activation.38,39 To test whether NF-κB inhibition could be monitored using our reporter system, cells expressing NF-Gluc were treated with 500 μM SSZ, a concentration reported to cause a 50% decrease in the binding of NF-κB to TRE elements. 39 As expected, we observed a twofold decrease in the Gluc basal level in response to SSZ treatment (Figure 3A). SSZ also produced a decrease in NFκB induction caused by TNF-α by fourfold (Figure 3B). A similar decrease in NF-κB induction caused by doxorubicin was also observed (data not shown). We further validated the inhibition of the TNF-α-mediated NF-κB activation by testing another NF-κB inhibitor, PTL, which blocks the IKK complex. 40 PTL almost abolished the activation of NF-κB by TNF-α, yielding a modest threefold increase in Gluc activity when combined with TNF-α (see Figure 3B). To determine the kinetics of the SSZ-mediated inhibition, we treated cells with either SSZ, TNF-α alone, or a combination of both TNF-α and SSZ. Aliquots of the cell-free conditioned medium were collected over time and assayed for Gluc activity. The inhibitory effect of SSZ on NF-κB was sustained up to 48 hours (Figure 3C). Further, a dose increase in SSZ in TNF-α-treated cells correlated with a decrease in Gluc activity (Figure 3D). In conclusion, the NF-Gluc reporter system proved to be a useful tool in monitoring both activation and inhibition of NF-κB in real time.
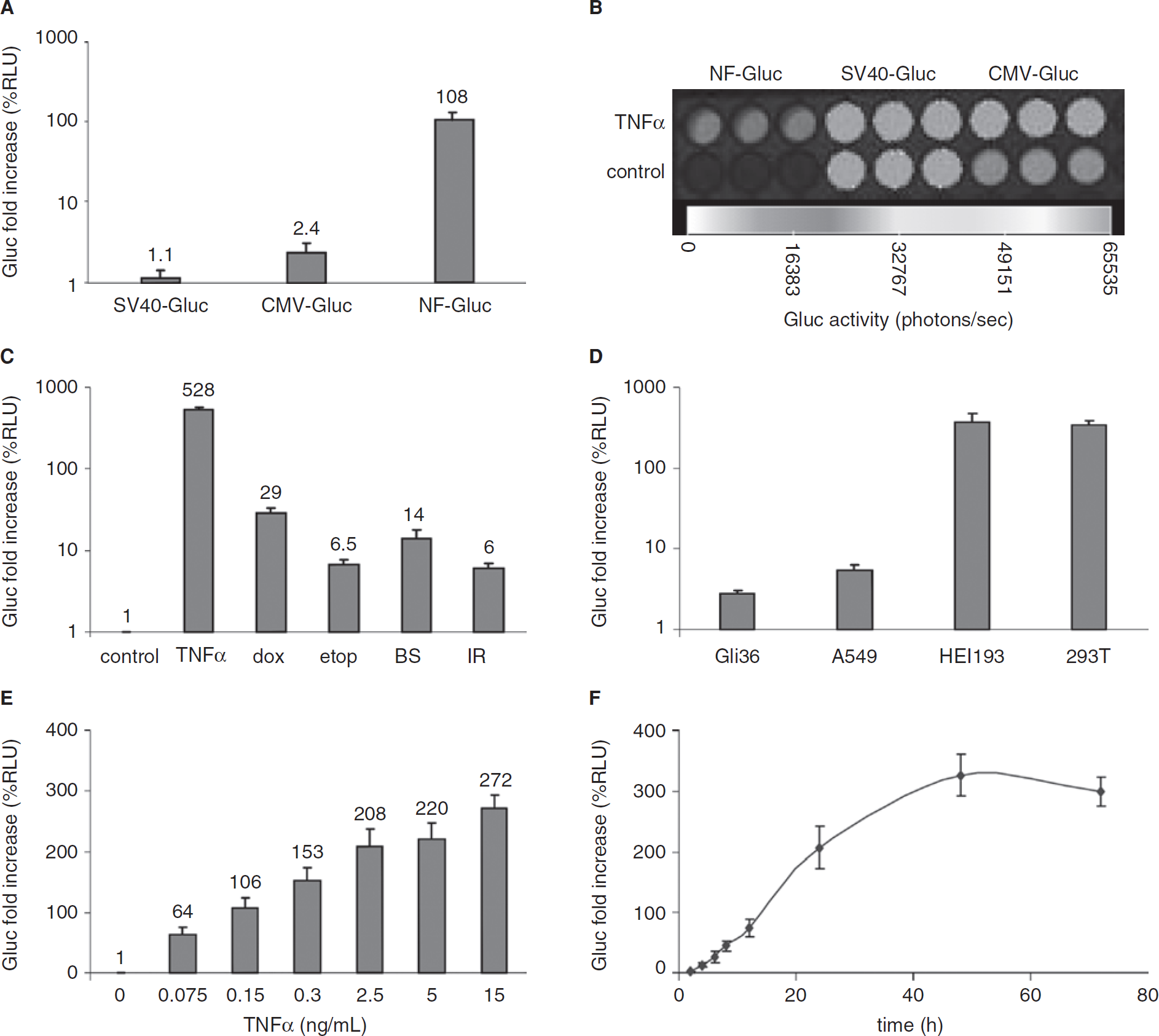
Monitoring of nuclear factor κB (NF-κB) activation in culture. A and B, 293T cells were transfected with a plasmid carrying the expression cassette for Gluc under control of the NF-κB, cytomegalovirus (CMV), or SV40 promoter and treated with tumor necrosis factor α (TNF-α) (10 ng/mL). Twenty-four hours later, aliquots of the conditioned medium were assayed for Gluc activity either using the luminometer (A) or the charge-coupled device camera (B) after the addition of coelenterazine. C, 293T cells stably expressing NF-Gluc were subjected to different treatments: TNF-α (20 ng/mL), etoposide (etop) (2 μM), doxorubicin (dox) (0.125 μM), bleomycin sulfate (BS) (50 μg/mL), or ionizing radiation (IR) (5 Gy). D, Different cell lines, Gli36, A549, HEI193, and 293T cells, were transduced with lenti-NF-Gluc and treated with TNF-α (20 ng/mL). E, 293T cells expressing NF-Gluc were treated with different concentrations of TNF-α. F, Time response curve for TNF-α induction. 293T cells expressing NF-Gluc were treated with TNF-α (2.5 ng/mL). In C to F, 15 μL aliquots of the conditioned medium were assayed for Gluc activity 24 hours posttreatment (C – E) or at different time points (F). Data are presented as the average fold increase in Gluc activity in treated cells compared with untreated cells ± SD (n = 4).
Next, we determined whether we could use the NF-Gluc reporter system to monitor the induction of NF-κB in a biologically relevant system. We decided to determine the induction of NF-κB in angiogenic endothelial cells, a well-studied process. 41 Low-passage HBMVEC-mCherry cells were transduced to express NF-Gluc. These cells formed a confluent monolayer and were viable for at least three passages, as determined by microscopic monitoring (Figure 4A). On coculturing of these cells on a Matrigel substratum with U87-Cerulean cells or in EBM containing an angiogenic cocktail (EGM), a tubule network was visualized using fluorescence microscopy (see Figure 4A). 42 The induction of endothelial tubules in vitro has been described to be partly mediated by NF-κB. 41 In parallel to the fluorescence microscopic analysis, we analyzed the conditioned medium from these cells for Gluc activity. A clear induction of the NF-κB-controlled Gluc signal was measured after stimulation of the HBMVECs with U87 glioma cells or the EGM medium containing a cocktail of angiogenic factors (Figure 4B), confirming a role for NF-κB in glioma angiogenesis in vitro.

Monitoring of nuclear factor κB (NF-κB) inhibition. 293T cells expressing NF-Gluc were plated in a 96-well plate and Gluc activity was assayed in an aliquot of conditioned medium 24 hours posttreatment with different drugs. A, Cells were treated with either phosphate-buffered saline (control) or sulfasalazine (SSZ) (500 μM). Data are presented as RLU/s ± SD (n = 4). B, Cells were treated with tumor necrosis factor α (TNF-α) (5 ng/mL) in the presence or absence of SSZ (500 μM) or parthenolide (PTL) (2 μM). C, Time course for NF-κB induction and inhibition after TNF-α (20 ng/mL) and/or SSZ (500 μM) treatment. Aliquots from the conditioned medium were assayed for Gluc at different time points posttreatment. D, Cells were treated with TNF-α (20 ng/mL) and different doses of SSZ. In B to D, data are presented as average fold change in Gluc activity compared with untreated samples ± SD (n = 4). *p ≤ .05, Student t-test.
In Vivo Blood Monitoring of NF-kB Activity
Recently, we showed that the level of Gluc in the blood of experimental animals bearing cells expressing this reporter under a constitutively active promoter can be used as a marker for cell viability and proliferation. 14 We therefore hypothesized that under the NF-κB TREs, the Gluc expression should reflect the physiologic state of NF-κB activation. Hence, we used our reporter construct for in vivo assessment of NF-κB activity by monitoring Gluc activity in the blood. HEI193 human schwannoma cells expressing either NF-Gluc or Fluc-mCherry as a control were implanted subcutaneously in nude mice. One week later, 10 μL aliquots of blood were collected and assayed for Gluc activity. In parallel, the Gluc activity in tumors was quantified by in vivo bioluminescence imaging using the CCD camera after intravenous injection of coelenterazine. Mice were then intravenously injected with 100 μL of TNF-α (16 μg/kg body weight) or PBS control, and blood was collected at different time points. An increase in Gluc activity in the blood was observed starting at 4 hours after TNF-α injection and reaching a maximum at 16 hours followed by a gradual decrease to the basal level 36 hours posttreatment (Figure 4C). In parallel, the mice were also imaged with the CCD camera for Gluc expression 12 and 48 hours after TNF-α injection. An increase in Gluc expression was also detected by in vivo bioluminescence imaging 12 hours posttreatment followed by a decrease to the basal level 48 hours later, which correlated with the Gluc-blood level (Figure 4D). To investigate whether the NF-Gluc reporter could be used to monitor NF-κB inhibition by SSZ in vivo, similar mice bearing xenografts of HEI193-NF-Gluc cells were treated with either SSZ (15 mg/kg body weight) or TNF-α and SSZ. A 2.5-fold decrease in the NF-κB basal level in blood was detected on SSZ treatment. Moreover, SSZ inhibited the NF-κB induction in HEI193 tumors after TNF-α treatment (see Figure 4C). To confirm these results in another cell type, we implanted subcutaneously 293T cells expressing either NF-Gluc or Fluc-mCherry as a control and treated mice with similar doses of TNF-α, SSZ alone, or a combination of both. Similar induction of Gluc expression in response to TNF-α-mediated NF-κB activation and a similar decrease in Gluc activity on treatment with either SSZ alone or TNF-α and SSZ was observed (data not shown).

Monitoring of nuclear factor κB (NF-κB) activation during tumor formation in vitro and in vivo. A, HBMVEC-mCherry cells infected with lenti-NF-Gluc were cultured under different conditions, as a monolayer on plastic (top left) or on Matrigel-coated plates in basal medium only (EBM) (top right) or basal medium supplemented with a cocktail of angiogenic factors (EGM) (bottom right), or with U87-CFP glioma cells (bottom left) (size bar = 300 μm). Images were obtained 24 hours post-culture. B, Twenty-four hours after culturing, Gluc activity was assayed in the conditioned medium. Data are presented as the average fold increase in Gluc activity compared with control monolayer culture ± SD (n = 4). *p ≤ .05, Student t-test. C, One million HEI193 cells were implanted subcutaneously in nude mice. One week later, mice were injected intravenously with either tumor necrosis factor α (TNF-α) (16 μg/kg of body weight), TNF + sulfasalazine (SSZ) (15 mg/kg of body weight), SSZ only, or phosphate-buffered saline (control). At different time points, 5 μL of blood was withdrawn and assayed for Gluc activity. D, Bioluminescence obtained from HEI193-NF-Gluc cells using a charge-coupled device (CCD) camera at time 0, 12, and 48 hours posttreatment. Data are presented as RLU/s ± SD (n = 5) with a CCD image of one representative mouse from each group.
Monitoring of NF-κB Activation in Monocytes
NF-κB plays a critical role in regulated expression of a large variety of genes involved in immune and inflammatory responses, such as cell adhesion molecules, chemokines, and cytokines. 43 Some cytokines, such as TNF-α, directly activate NF-κB to amplify the primary inflammatory response. Monitoring of NF-κB activation in vivo may contribute to a better understanding of the innate immune response activation. We transduced U937 human leukemic monocyte lymphoma cells with lenti-NF-Gluc. Initially, these cells were exposed to different amounts of TNF-α in culture. At different time points, aliquots of the conditioned medium were assayed for the Gluc activity. A dose-dependent increase in Gluc expression in the treated U937 cells compared with nontreated cells indicated that NF-κB was activated by up to eightfold (24 hours posttreatment) in response to TNF-α (Figure 5A). To corroborate these findings in an in vivo model, U937 cells expressing NF-Gluc were injected intraperitoneally, and 1 hour later, mice were injected with either PBS or 80 μg/kg body weight of TNF-α in a similar route. Before TNF-α injection and at different time points after injection, 5 μL aliquots of blood were withdrawn and assayed for Gluc activity. The Gluc blood level measured in response to NF-κB activation showed a maximum at 24 hours post-TNF-α injection (eightfold), after which the signal decreased to the basal level (48 hours later), suggesting a transient induction of NF-κB activation. At no time point did the PBS control group show any change in Gluc blood activity (Figure 5B). Also, prior to TNF-α injection and 24 hours postinjection, mice were imaged with the CCD camera after intravenous injection of coelenterazine. At neither time point was there any positive signal, with the CCD camera supporting the high sensitivity of the Gluc blood assay compared with in vivo bioluminescence imaging in monitoring NF-κB activation in dispersed monocytes (data not shown). The NF-Gluc reporter and the Gluc blood assay provide a means to monitor NF-κB activation in immune cells, which can serve as a tool to study the dynamics of immune activation in subsets of immune cells in small animals.

Monitoring of nuclear factor κB (NF-κB) activation in monocytes. A, U937 cells expressing NF-Gluc were treated with different concentrations of tumor necrosis factor α (TNF-α). At different time points, the Gluc activity was assayed in 10 μL of conditioned medium. B, U937-NF-Gluc cells were injected intraperitoneally, and 1 hour later, mice were injected with either phosphate-buffered saline (control) or TNF-α (80 μg/kg of body weight) in the same route. Before and at different time points after treatment, Gluc activity was monitored in 20 μL of blood. Data shown are average RLU/s ± SD (n = 6).
Dual Blood Monitoring of NF-κB Activity and Cell Growth during Tumor Development
Given that NF-κB is involved in tumor progression, 12 we sought to look for NF-κB activation during tumor development. Gli36 human glioma cells were cotransduced with lenti-NF-Gluc, lenti-Fluc-mCherry, and lenti-SEAP and implanted subcutaneously in mice. Before and at different time points postimplantation, serum was collected and assayed for Gluc or SEAP activity as an index for NF-κB activation and cell growth, respectively. An increase in Gluc signal and therefore NF-κB activation was observed over time, which correlated with an increase in SEAP signal, a marker for tumor cell proliferation (Figure 6A). In parallel, 5 μL of urine was collected and assayed for Gluc activity, which showed a similar increase in Gluc expression, proving that the Gluc level in urine can also be used as an index of NF-κB activation (Figure 6B). In another experiment, mice were implanted with different amounts of these cells. One week later, tumor volume was monitored using in vivo Fluc bioluminescence imaging after intraperitoneal injection of D-luciferin and serum was assayed for Gluc or SEAP activity. The Gluc level in serum as a marker for NF-κB activation correlated with the serum SEAP level, which, in turn, correlated with tumor volume as assessed by in vivo bioluminescence imaging (Figure 6C). On the other hand, when these tumor cells were injected intravenously, an increase in Gluc value (7- to 10-fold) was observed in serum 2 hours postinjection, indicating an immediate activation of NF-κB in these cells, which then dropped back to the basal level (near SEAP signals) 18 hours later (Figure 6D).

Dual monitoring of tumor formation and nuclear factor κ (NF-κB) activation. One million Gli36 human glioma cells expressing Fluc, NF-Gluc, and SEAP were implanted subcutaneously in nude mice. A and B, At different time points, blood or urine was withdrawn and serum was assayed for either Gluc or SEAP activity (A) and urine for Gluc activity (B). C, One week postimplantation, tumor volume was imaged with in vivo Fluc bioluminescence imaging using the charge-coupled device camera, and Gluc and SEAP serum levels were assayed as in A. D, Gli36 cells expressing NF-Gluc and SEAP were injected intravenously, and the serum Gluc and SEAP activity was monitored at different time points as in A.
Discussion
We developed a novel NF-κB reporter system based on the naturally secreted Gaussia princeps luciferase.14,17 The NF-Gluc reporter proved to be a useful tool for sensing both NF-κB activation and inhibition in different models, including tumors, angiogenesis, and inflammation. Moreover, the Gluc reporter allowed monitoring of NF-κB activity by measuring its level in an aliquot of the conditioned medium of cultured cells or in the blood or urine of animals at sequential time points. Further, in the context of concentrated local NF-Gluc reporter in vivo, the Gluc signal can be confirmed and localized using in vivo bioluminescence imaging.
NF-κB is a ubiquitous transcription factor that plays a critical role in regulating expression of a large variety of genes involved in immune and inflammatory responses, such as cell adhesion molecules, chemokines, and cytokines, but also genes controlling other biologic processes, such as cell survival, apoptosis, and differentiation. Activation of this transcription factor is associated with several physiologic disorders, notably inflammation and cancer. Monitoring of NF-κB activation may contribute to a better understanding of such biologic processes and pathologic conditions. Several reporter systems have been used to study the expression and activation of NF-κB. Previous studies have used NF-κB responsive elements driving the expression of LacZ,44,45 Fluc, 46 enhanced green fluorescent protein (eGFP 47 ), or SEAP 48 reporter genes. Although these systems proved to be useful in detecting NF-κB activation, they may each have several disadvantages in the context of in vivo analysis and sequential monitoring of NF-κB activity compared with the NF-Gluc reporter developed here. Fluc requires D-luciferin injection before imaging; thus, a total clearance of the substrate is essential before the next imaging session can be started, complicating the monitoring of NF-κB activation kinetics, unless using a pump system that continuously delivers the substrate to the animal. 49 In addition, anesthetizing the animals can be a major limitation during kinetic studies, requiring multiple imaging sessions over a short period of time or during studies in which the animal needs to stay awake. The eGFP reporter gene is partly compromised by its relatively low sensitivity and in most cases requires animal sacrifice before fluorescent analysis or, alternatively, the use of more elaborate techniques, such as epifluorescence microscopy. A high signal to noise ratio owing to autofluorescence in many organs decreases dramatically the sensitivity of in vivo fluorescence imaging. LacZ staining requires animal sacrifice and tissue sectioning before analysis. SEAP overcomes most of these problems; however, we recently showed that this reporter is 20,000-fold less sensitive than Gluc in cultured cells, with a linear range with respect to cell number covering less than three orders of magnitudes. 50 Further, the SEAP assay requires several different incubation steps before analysis, making this assay more laborious and time consuming. On the other hand, the Gluc blood/urine assay is simple and short, requiring only the addition of its substrate coelenterazine and luminometer analysis, and has a linear range of over five orders of magnitude with respect to cell number. 14
The NF-κB transcription factor is known to be activated in many cancer types, including lung and ovarian cancer, astrocytomas, melanoma, prostate adenocarcinoma, and glioblastoma, and was shown to correlate with disease progression.6,7,32–34,36,37,51 Therefore, this reporter could be used to identify and monitor potential chemotherapeutics based on interference or potentiation of NF-κB activity in vitro and in vivo. Given that NF-κB has been shown to be involved in tumor angiogenesis and invasiveness, 12 the NF-Gluc reporter can also be used as a sensor for tumor angiogenesis. The neovascularization process occurs through a series of organized steps, which is initiated by remodeling of the extracellular matrix. NF-κB plays a major role in this remodeling process by inducing expression of various genes such as vascular endothelial growth factor, plasminogen activator inhibitors, and matrix metalloprotein ases in endothelial cells.52–54 Further, NF-κB plays a major role in the migration and invasion of endothelial cells55,56; therefore, the NF-Gluc reporter could be used to determine the kinetics of NF-κB activation during the various steps of angiogenesis.
Besides cancer, identifying and testing of novel drugs that interfere with NF-κB activity can be important for the treatment of a number of disorders, including neurologic disorders, 57 rheumatoid arthritis, 58 inflammatory bowel disease, 59 and asthma. 60 The NF-Gluc reporter may also prove useful for high-throughput drug screening for NF-κB activators and inhibitors, which subsequently can be validated in vivo using the NF-Gluc-blood assay. Not many systems are available such as NF-Gluc, allowing both in vitro screening and kinetic analysis, as well as in vivo validation of a relatively large number of drugs in a short period of time. Moreover, this reporter system is easy to implement, is cost and time-effective, and can be extended to study other physiologically regulated transcription elements, such as cyclic adenosine monophosphate, p53, ISRE, TARE, and SRF, making it a versatile tool for studying transcriptional activation in vitro and in vivo in a noninvasive and quantitative manner.
Footnotes
Acknowledgments
We would like to thank Dr. Ralph Weissleder (Center for Molecular Imaging Research, Massachusetts General Hospital) for the use of the CCD camera and Dr. Xandra Breakefield for her insights.
Financial disclosure of authors: This work was supported partially by grants from the National Institutes of Health/National Cancer Institute (P50 CA86355-07) and the VONK-SEMMY Foundation.
Financial disclosure of reviewers: None reported.