
Abstract
Low-density lipoprotein (LDL) provides a highly versatile natural nanoplatform for delivery of visible or near-infrared fluorescent optical and magnetic resonance imaging (MRI) contrast agents and photodynamic therapy and chemotherapeutic agents to normal and neoplastic cells that overexpress low-density lipoprotein receptors (LDLRs). Extension to other lipoproteins ranging in diameter from about 10 nm (high-density lipoprotein [HDL]) to over a micron (chylomicrons) is feasible. Loading of contrast or therapeutic agents onto or into these particles has been achieved by protein loading (covalent attachment to protein side chains), surface loading (intercalation into the phospholipid monolayer), and core loading (extraction and reconstitution of the triglyceride/cholesterol ester core). Core and surface loading of LDL have been used for delivery of optical imaging agents to tumor cells in vivo and in culture. Surface loading was used for delivery of gadolinium-bis-stearylamide contrast agents for in vivo MRI detection in tumor-bearing mice. Chlorin and phthalocyanine near-infrared photodynamic therapy agents (≤ 400/LDL) have been attached by core loading. Protein loading was used to reroute the LDL from its natural receptor (LDLR) to folate receptors and could be used to target other receptors. A semisynthetic nanoparticle has been constructed by coating magnetite iron oxide nanoparticles with carboxylated cholesterol and overlaying a monolayer of phospholipid to which apolipoprotein A1 or E was adsorbed for targeting HDL or adsorbing synthetic amphipathic helical peptides ltargeting LDL or folate receptors. These particles can be used for in situ loading of magnetite into cells for MRI-monitored cell tracking or gene expression.
LIPOPROTEINS are natural nanoparticles spanning a range of diameters from about 10 nm (high-density lipoprotein [HDL]) to over a micron [chylomicrons]). 1 All lipoproteins contain a phospholipid monolayer shell and a hydrophobic lipid core consisting of triglycerides and cholesterol esters. Many contain protein components adsorbed onto the phospholipid monolayer. The most commonly studied lipoprotein, low-density lipoprotein (LDL), which is ≈20 nm in diameter, contains a very large protein component, apolipoprotein B-100 (550 kDa, 4,536–amino acid residues), that covers about 40% of its surface area and targets the particle to low-density lipoprotein receptors (LDLRs) in a number of normal tissues, such as liver, adrenal glands, and ovaries. LDL and HDL transport cholesterol to and from tissues and play pathologic and protective roles, respectively, in cardiovascular disease. Because a number of tumors overexpress LDLRs, this particle has been used as a delivery vehicle for a number of lipophilic drugs. 2 Our laboratories have been developing LDL and, more recently, other lipoproteins for delivery of optical and nuclear magnetic resonance probes, as well as photodynamic therapy (PDT) agents and cancer drugs, to solid tumors. Here we describe the current status of this research.
Methods
Preparation of Lipoproteins
Methods for preparation of LDL and chemically modified LDL have been previously described. 3 For preparation of the iron oxide–encapsulating lipoproteins, iron (III) acetylacetonate was added to phenyl ether with 1,2-hexadecanediol and cholic acid under nitrogen and then heated to 265°C to form cholesterol-coated iron oxide particles. The collected oily particles were dispersed in chloroform, to which phosphatidylcholine was added, and the solution was dried. Phosphate buffered saline including ethylenediaminetetraacetic acid and NaN3 was added to the dried sample and sonicated at 50°C to form oil in water phospholipid micelles containing a shell of phospholipid and a core of cholesterol-coated iron oxide particles. Apolipoprotein A-I was finally added to the micelle solution under sonication to decorate the phospholipid micelle, forming lipoprotein particles with a total diameter of 20 nm.
Synthesis of Near-Infrared Fluorophores and Magnetic Resonance Imaging Contrast Agents
Cell Lines and Animal Models
Confocal Microscopy
Redox Scanning
These methods have been previously described. 4
Optical Imaging
Imaging was performed with a Xenogen imaging system (Palo Alto, CA) under isoflurane anesthesia.
Magnetic Resonance Imaging
These methods have been previously described. 6
Results and Discussion
Lipoproteins: Nature's Nanoparticles
LDL is a 22 nm particle (density 1.006–1.063 g/mL) that contains one copy of apolipoprotein B-100 that is rich in amphipathic α-helices. 6 The hydrophobic faces of these helices are adsorbed onto the outer surface of the phospholipid monolayer, which also contains variable amounts of free cholesterol. The inner surface of the monolayer is adsorbed onto a lipid core consisting of cholesterol esters (≈1,500 per particle) and triglycerides.
The mechanism of internalization of LDL in cells was delineated through the classic studies of Brown and Goldstein. 7 Through electrostatic interactions between highly cationic receptor binding sequences on apolipoprotein B-100 with complementary anionic sequences on the cell surface, the particle binds to LDLRs embedded in clathrin-coated pits on the cell surface. The receptor-LDL complex is internalized by endocytosis and digested in lysosomes by acid hydrolysis and enzymatic breakdown into free cholesterol, fatty acids, and amino acids; the receptors are cycled back to the cell surface in about 10 minutes. The process can then be repeated with another LDL particle. The turnover time for cell surface LDLRs is about 24 hours. Any probes or drugs incorporated into the LDL particle accumulate within the targeted cell, providing a very effective amplification mechanism for drug and imaging probe delivery.
A number of normal tissues, such as liver, adrenal glands, and ovaries, contain high levels of LDLRs. Many malignancies also overexpress these receptors, including acute myelogenous leukemia (3- to 100-fold compared with normal cells), colon cancer (6-fold), adrenal adenoma (8-fold), and pancreatic, lung, brain, and prostate cancer. 8 Among the laboratory models of human cancer, B16 melanoma and HepG2 hepatoma are rich in LDLRs.9,10
Three methods have been used to bind imaging or therapeutic agents to LDL: protein loading, core loading, and surface loading. 2 Each has advantages and disadvantages; we have used each of these methods in our studies. Surface loading was used for delivery of near-infrared fluorophores (NIRFs) 4 and paramagnetic gadolinium (Gd)-chelated magnetic resonance imaging (MRI) probes,6,11 core binding for delivery of NIRFs and PDT agents,3,5 and protein loading for redirecting LDL to receptors other than LDLRs. 3 These applications are described below after a short description of the loading procedures.
The first method, protein loading, is performed by covalent attachment to side chains of apolipoprotein B-100, usually the amino groups of lysine or the phenol group of tyrosine. A number of investigators have attached chelating groups to lysine side chains and used these to bind 111In or 99mTc for single-photon emission computed tomography (SPECT)12,13 or 68Ga for positron emission tomography (PET). 12 Attachment of 18F-containing ligands to the lysine-ε-amino groups has also been used for PET. 14 Radio-iodination of tyrosine side chains for SPECT detection has been attempted but leads to modification of the transport properties of the protein. 15 The advantage of protein labeling is that stable products are formed. The disadvantage is that covalent binding may modify the delivery characteristics of LDL since the receptor binding site includes reactive lysines (see below).
The second method of LDL labeling is core labeling that introduces lipophilic probes into the core of the particle. Krieger and colleagues demonstrated that in the presence of starch, the lipid core of LDL can be extracted with a nonpolar solvent such as heptane while keeping the phospholipid and protein “shell” intact. 16 The lipophilic probe is introduced into the extract, the solvent is evaporated in vacuo, and the particle spontaneously reassembles with LDL protein recoveries of over 50%. This provides a very stable vehicle for delivery of lipophilic probes to cells via LDLRs.
The third method of LDL labeling is surface labeling, which can be used to noncovalently bind probes to the phospholipid surface of the phospholipid monolayer. In general, the probe is conjugated to one or two long hydrophobic chains and sometimes also to a cholesterol group. These entities intercalate between the phospholipid fatty acids and may even protrude into the lipid core (thereby anchoring the ligand within the particle). Very high yields have been achieved with fluorescent4,5 or paramagnetic probes.6,11 The method is easy to implement but prone to high leakage rates. The reason for this is that transfer of the surface probe to the outer phospholipid layer of the cell plasma membrane is thermodynamically favorable. Hence, substantial delivery of probes to cells could occur by pathways not involving receptor delivery.
LDL Surface and Core Loading of NIRFs/PDT Agents
Krieger and colleagues used a cholesterol laurate scaffold for surface binding of the fluorophore pyrene to LDL. 17 We used the same strategy to attach tricarbocyanine4,18 and tris[(porphinato)zinc(II)] fluorophores 19 to LDL. Figure 1A describes the structure of the tricarbocyanine near-infrared (NIR) dye IRD41 (LI-COR Biotechnology Division, Lincoln, NE) conjugated to an amino group on cholesterol laurate via a thiourea linkage. Cholesterol oleate conjugates were prepared similarly as well as pyropheophorbide cholesterol esters (Pyro-CE; Figure 1B). 5 The tricarbocyanine conjugates are charged, facilitating surface loading (although core loading is also feasible), whereas the Pyro-CE is neutral and most suitable for core loading. Pyropheophorbide is both a NIRF and a PDT agent.
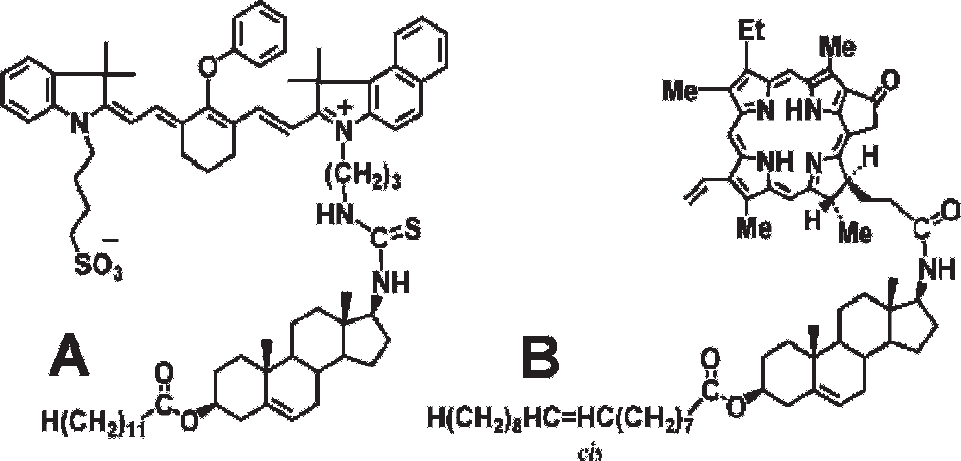
Structure of cholesterol ester conjugates with tricarbocyanine (A) and pyropheophorbide (B).
Delivery of both types of LDL-NIRF conjugates to B16 melanoma and HepG2 tumor cells was confirmed by confocal microscopy.4,5 Intense NIR fluorescence distributed uniformly throughout the cytoplasm demonstrated internalization of the NIRFs in the cytoplasm after digestion of the particles in the lysosomes. Excess unlabeled LDL blocked delivery, confirming that the dyes entered the cell via LDLRs.
Reconstituted low-density lipoprotein (r-LDL) core labeled with Pyro-CE was injected into the tail veins of nude mice with subcutaneous B16 or HepG2 tumors. Analysis of blood specimens indicated clearance of the r-LDL in ≈30 minutes (data not shown). Localization of the dye in the tumor was monitored by cryospectrophotometry using reflectance fluorescence measurements by the method of Quistorff and colleagues (“redox scanning”). 20 Images were taken of 10 μm slices with an isotropic inplane resolution of 100 μm. Figure 2 shows that in the B16 melanoma, the dye accumulated mostly in the periphery of the tumor. Large areas of central necrosis were virtually devoid of label. In the hepatoma, much more intense binding was noted in the center of the tumor, and there was much less necrosis. Scatchard analysis of binding of LDL surface labeled with the commercial visible monocarbocyanine dye DiI to isolated B16 and HepG2 cells indicated that both tumors exhibited similar numbers of LDLRs per cell, but the affinities of the HepG2 receptors were about twice those of the B16 receptors. 4 However, the distribution of Pyro-CE LDL appears to be determined primarily by the perfusion properties of these tumors, with the melanoma exhibiting a well-perfused periphery and a poorly perfused central region, whereas perfusion of the hepatoma was probably more uniform.
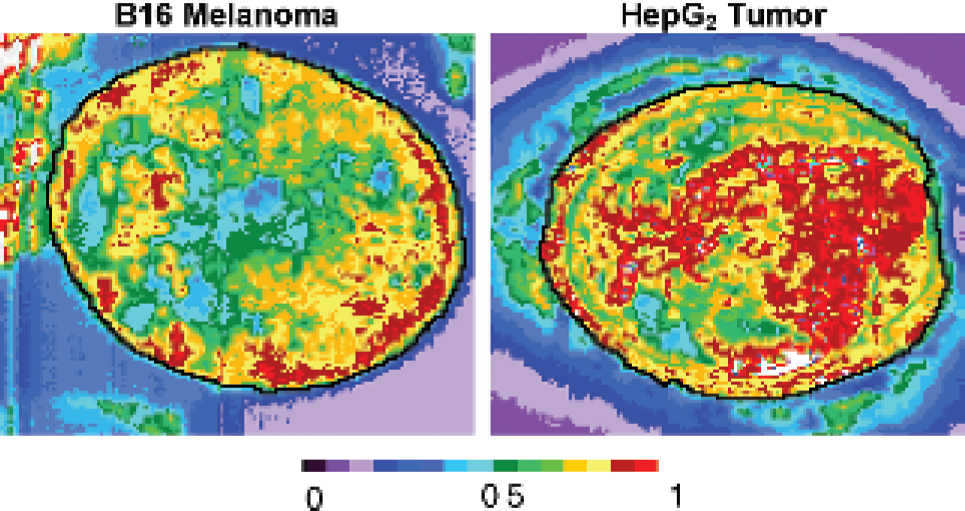
Redox scans of single slices of B16 melanoma and HepG2 hepatoma after intravenous injection of the reconstituted low-density lipoprotein–pyropheophorbide–cholesterol ester complexes into mice with subcutaneous tumors.
A preliminary study was performed comparing NIR images of tumor-bearing mice injected with surface-loaded and core-loaded LDL particles. The commercial NIR tricarbocyanine DiR was used to label the LDL. Figure 3 shows much more extensive and nonspecific binding of the surface-loaded dye. Therefore, whenever possible, core loading should be used to label LDL.

Preliminary comparison of the effects of surface- and core-loaded low-density lipoprotein (LDL) on distribution of the dye delivered by intravenous injection of LDL labeled with DiR.
Surface Binding of Gd Chelates for MRI
For MRI detection, the Gd–diethylenetriamine pentaacetic acid (DTPA)–bis-stearylamide (SA) complex, a bidentate phospholipid intercalator, was synthesized and conjugated to LDL by surface loading (Figure 4). 6 In this instance, surface loading was required since relaxation enhancement occurs by direct contact of the Gd with water. Between 100 and 500 Gd-DTPA-SA complexes could be conjugated to the LDL surface. The optimum relaxivity was achieved with 180 Gd complexes per LDL. Table 1 compares the relaxivities of commercial Gd-diamide MRI contrast agent with those of the isolated Gd-DTPA-SA and its complex with LDL (180 Gd/LDL particle). Similar relaxivities were obtained with the isolated chelates, but the complex of the nanoparticle with 180 Gd complexes produced much higher relaxation enhancement.
Relaxivity (mM−1s−1) of Gadolinium Analogues at 4.7 T (21°C)

Gd = gadolinium; Gd-DTPA-SA = gadolinium–diethylenetriamine pentaacetic acid–bis-stearylamide; LDL = low-density lipoprotein.
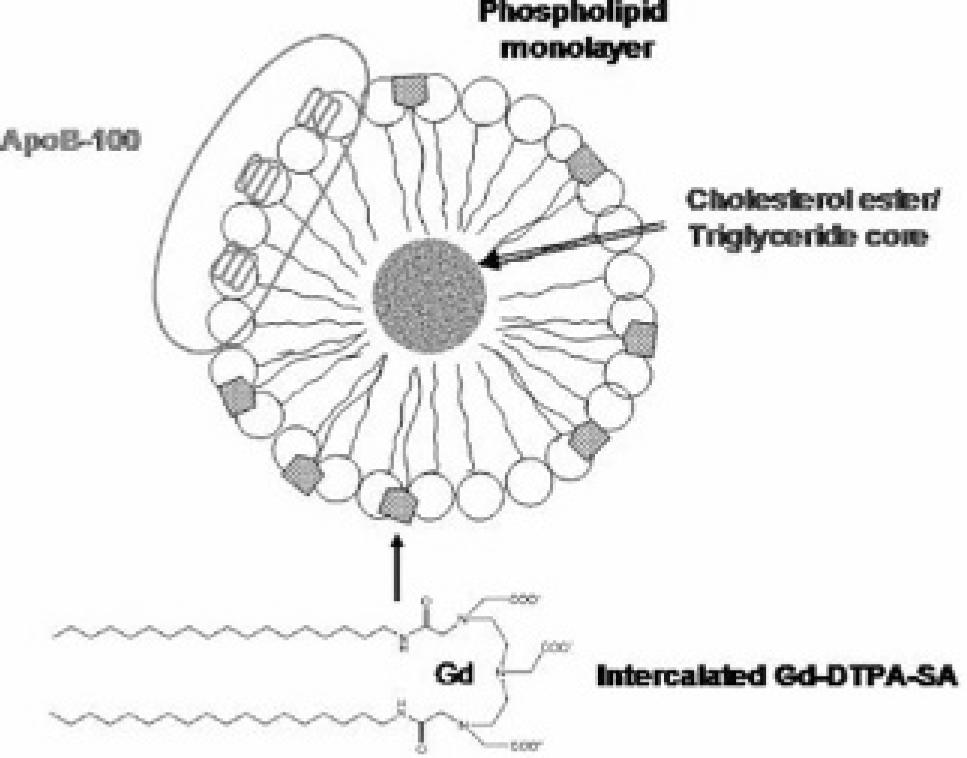
Schematic representation of surface loading of low-density lipoprotein with gadolinium–diethylenetriamine pentaacetic acid–bis-stearylamide (Gd-DTPA-SA) complex for magnetic resonance imaging detection. ApoB-100 = apolipoprotein B-100.
Figure 5 compares the relaxation enhancement of liver and tumor in axial MRIs of a mouse with a subcutaneous HepG2 hepatoma xenograft at various times after intravenous injection of the Gd-DTPA-SA-LDL complex. The enhancement of the tumor becomes significant 24 hours postinjection.
These studies represent initial attempts at using LDL as a vehicle for delivery of Gd. Substantially higher relaxivities can be achieved with other Gd chelates, particularly with those containing two water ligands per Gd atom. Further studies are being pursued to optimize the Gd enhancements induced by agents conjugated with LDL. It must be noted that these particles will be degraded on cellular incorporation, and the identity and properties of the intracellular Gd complex are as yet undetermined.
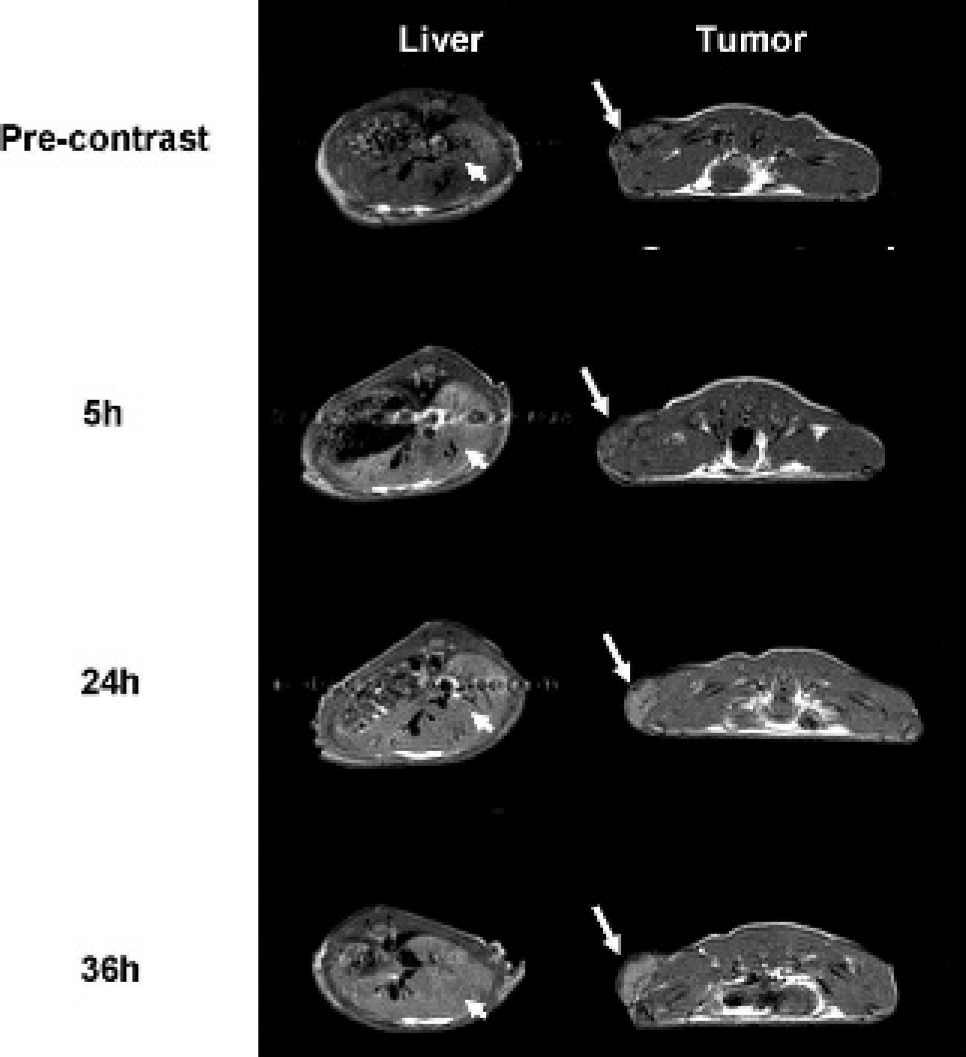
Comparison of T1-weighted magnetic resonance image of a nude mouse with a subcutaneous HepG2 tumor before injection of gadolinium–diethylenetriamine pentaacetic acid–bis-stearylamide–low-density lipoprotein (180:1; 0.04 mmol/kg) (top), 5 hours after injection (liver 55% enhancement, tumor 9%), 24 hours after injection (liver 25% enhancement, tumor 25%), and 36 hours after injection (liver ≈25% enhancement, tumor 25%). Figure reprinted from Corbin IR et al 6 by permission of Neoplasia.
Receptor-Targeted PDT Agents
PDT agents or photoactivators are highly promising new therapeutic agents that are currently used to treat superficial malignancies, macular degeneration, or metastasized malignancies accessible by endoscopy or surgical exposure. 21 These agents are administered in an inactive form but produce highly cytotoxic reactive singlet oxygen or superoxide anions ( 1 O2−) when activated by specific absorption wavelengths of visible or NIR light. The agents are currently administered in an untargeted form and are used either to ablate blood vessels or to destroy mitochondrial activity when they enter the cell.
Their mechanism of action is well defined. Absorption of a photon of light leads to transition of the photoactivator from its stable S0 ground state to an excited singlet state (S1) with typical lifetimes of ≈10−6 seconds. Fluorescent molecules undergo similar excitation and decay back to the ground state, emitting light of slightly lower energy than the absorbed photon. However, photoactivators undergo intersystem crossing to an excited triplet state (T1) with much longer lifetimes (≈10−2 seconds). With phosphorescent agents, emission of photons occurs from the triplet state to the ground state. However, photoactivators undergo two types of reactions from the T1 state with oxygen molecules, a type I reaction involving electron transfer to form a superoxide (or singlet oxygen radical anion, 1 O2−) or a type II reaction involving a collision with molecular oxygen ( 3 O2) that also produces singlet oxygen. The singlet oxygen radical anion is highly reactive with water, producing hydroxyl radicals that alkylate deoxyribonucleic acid (DNA) and various essential proteins.
Chlorophyll a and bacteriochlorophyll are magnesium chelates that undergo such photoactivation reactions in the NIR range. These agents are of little clinical utility because they are highly unstable and undergo self-degradation by auto-oxidation, enolization, photobleaching, transesterification, and demetalation. A variety of derivatives of these chlorin molecules have been produced, some of which are much more stable and useful for clinical PDT.22,23
Initial incorporation of these photoactivators into LDL was achieved by attaching a hydrophobic cholesterol oleate group or a bis-oleic acid group to bacteriochlorophyll (BCh), producing BChl-CE and BChl-BOA, respectively. BChl-CE absorbs at 748 nm and emits at 762 nm, which are suitable for in vivo activation of tissues at substantial depths (3–8 cm) from the surface. The key limitation of these derivatives, besides their intrinsic instability, is their low core-loading efficiency into LDL (< 50:1).
Phthalocyanines (Pcs) are planar porphyrin photoactivators that absorb and emit photons in the NIR, making them suitable for clinical application. Planar Pcs aggregate, which impedes core loading into LDL and leads to self-quenching. However, incorporation of a silicon atom in the center of the Pc permits introduction of axial ligands that sterically hinder aggregation and facilitate core loading of planar analogues into LDL. The optical properties of these agents are also suitable for NIR activation. One of these agents, Pc4 with sialobutylamino and OH axial ligands, is currently in a National Cancer Institute phase I trial. A Pc with bisoleate axial ligands and t-butyl peripheral ligands (SiPc-BOA) was produced at a relatively high yield (50%) with excitation (Ex) 684, emission (Em) 692 (excitation and emission wavelengths, respectively), and high solubility in nonpolar solvents. 24 This derivative was readily core-loaded into LDL (55–70% yield) with high payloads (400:1) and minimal change in particle dimension determined by electron microscopy (r-SiPc-BOA-LDL: diameter 23.2 ± 4.6 nm, n = 30 vs LDL: diameter 20.0 ± 2.7 nm, n = 37). Specificity for LDLR was confirmed by confocal microscopy with HepG2 cells. Preliminary in vitro studies indicate that ≈90% cell kill of HepG2 cells treated with SiPc-BOA-LDL (5.8 μg/mL) irradiated at a fluence rate of 5 J/cm. 24
Rerouting of LDL to Folate Receptors
High levels of LDLR in various normal tissues, particularly liver and the adrenal glands, limit the specificity of LDL-based nanoparticles for tumor cells. Given that a number of much more tumor-specific receptors have been identified, we sought a method to redirect the LDL nanoparticles to these alternative targets. Our strategy was to alkylate the lysine ε-amino side chains with appropriate receptor-targeting ligands. To demonstrate the principle, we chose folic acid (FA) receptors (FRs) since these are overexpressed in a number of tumors, particularly ovarian. Key to the success of this strategy is the lower pKa (8.9) and, hence, higher sensitivity to alkylation of the lysine amino side chains of the LDLR-targeting region of LDL. 25 Figure 6 shows that five of the eight amino acids of the receptor binding sequence are cations; similarly, apolipoprotein E, which also targets LDLRs, has six of eight cationic amino acids at its receptor binding site. Therefore, alkylation of the lysine side chains of LDL rapidly eliminates LDLR binding activity at the same time as the addition of FA ligands to these amino groups redirects the particle to FRs. Alkylation of 20% of the lysine side chains totally abolishes LDLR binding ability. 25
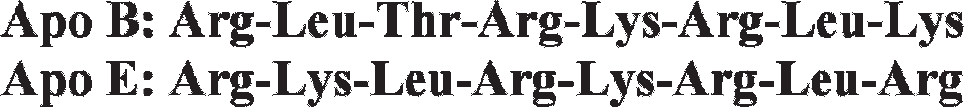
The low-density lipoprotein receptor binding sequences of apolipoprotein B-100 (ApoB) and apolipoprotein E (ApoE) contain clusters of cationic amino acids.
Apolipoprotein B-100 contains 357 lysine side chain amino groups, of which 225 are exposed and 132 are buried. Among the exposed lysines, there are 53 active lysine amino groups (pKa 8.9), with the rest exhibiting normal pKas (10.5) and normal reactivity. 3 The presence of clusters of cationic amino acids in the backbone sequence could account for all or some of these anomalously acidic amino side chains; however, regions of low dielectric constant could also facilitate deprotonation of some of the charged ammonium groups.
We attached folate groups to 170 or 47.6% of the lysine side chains of LDL, which totally abolished binding to LDLR (see below). 3 To enable confocal microscopy, we surface-loaded the commercial visible dye DiI (Invitrogen Corp., Carlsbad, CA) to the folate-conjugated LDL. The molar ratio of the DiL:LDL:FA was 55:1: ≈150 to 200; the diameter of the particle measured by electron microscopy was 26.1 ± 3.0 nm, which is slightly more than native LDL (see above). We also prepared LDL-FA particles missing legend under the eighth plate above.
We also prepared LDL-FA particles core loaded with the silicon phthalocyanine PDT agent (r-SiPc-BOA:LDL: FA = 1,500:1: ≈150 to 200; 24.0 ± 4.3 nm). The latter were adequately fluorescent for confocal microscopy.
Confocal microscopy studies of cell lines with specific types of receptors confirmed receptor-mediated delivery of DiI-LDL-FA to FRs and the absence of uptake by LDLR. 3 Briefly, KB nasopharyngeal carcinoma cells (FR+) internalized DiI-LDL-FA; the addition of excess FA abolished uptake, but the addition of excess unlabeled LDL did not. Chinese hamster ovary (CHO) cells (FR−) and HT1080 cells (FR−) failed to take up DiI-LDL-FA, as did HepG2 cells (FA−, LDLR+), but the latter bound DiI-LDL. Flow cytometry demonstrated a monotonic increase in fluorescence intensity of KB cells incubated with increasing concentrations of DiI-LDL-FA, but FA competitively inhibited uptake of the DiI-LDL-FA by these cells. Similar studies were performed with r-SiPc-BOA-LDL-FA. In KB (FR+) cells that internalized r-SiPc-BOA-LDL-FA, excess FA inhibited uptake, but excess unmodified LDL did not. CHO cells (FR−) did not internalize the r-SiPc-BOA-LDL-FA. HepG2 (FA−LDLR+) cells did not take up r-SiPc-BOA-LDL but did accumulate r-Pc-BOA-LDL.
Preliminary in vivo tumor localization studies were performed on mice with a subcutaneous HT1080 (FR−) tumor on one thigh and a KB (FR+) tumor on the other thigh. 26 At t = 0, 0.77 μm LDL-FA surface-loaded with the NIRF DiR (Ex: 748 nm, Em: 780 nm; DiR:LDL:FA = 8:1:105) was injected intravenously into the tail vein. Xenogen images (Ex: 710–760 nm, Em: 810–875 nm) measured at various times postinjection are shown in Figure 7. Five minutes after injection, the dye was distributed throughout the animal and in both tumors. At 2 hours postinjection, the dye had cleared from the FR− HT1080 tumor but was retained in the FR+ KB tumor. However, the bulk of the dye remained outside the tumor, particularly in the liver. At 24 hours, fluorescent intensity had increased substantially in the KB tumor and almost disappeared from the abdomen. The study demonstrates that the LDL particle can diffuse through leaky blood vessels into both tumors but is retained and accumulates in the tumor that contains the FRs. There also appears to be slow transfer of the nanoparticles between binding sites in the abdomen and in the tumor. The mechanism underlying this transfer requires further study. However, the data demonstrate that redirection of the particle to the FR has taken place.
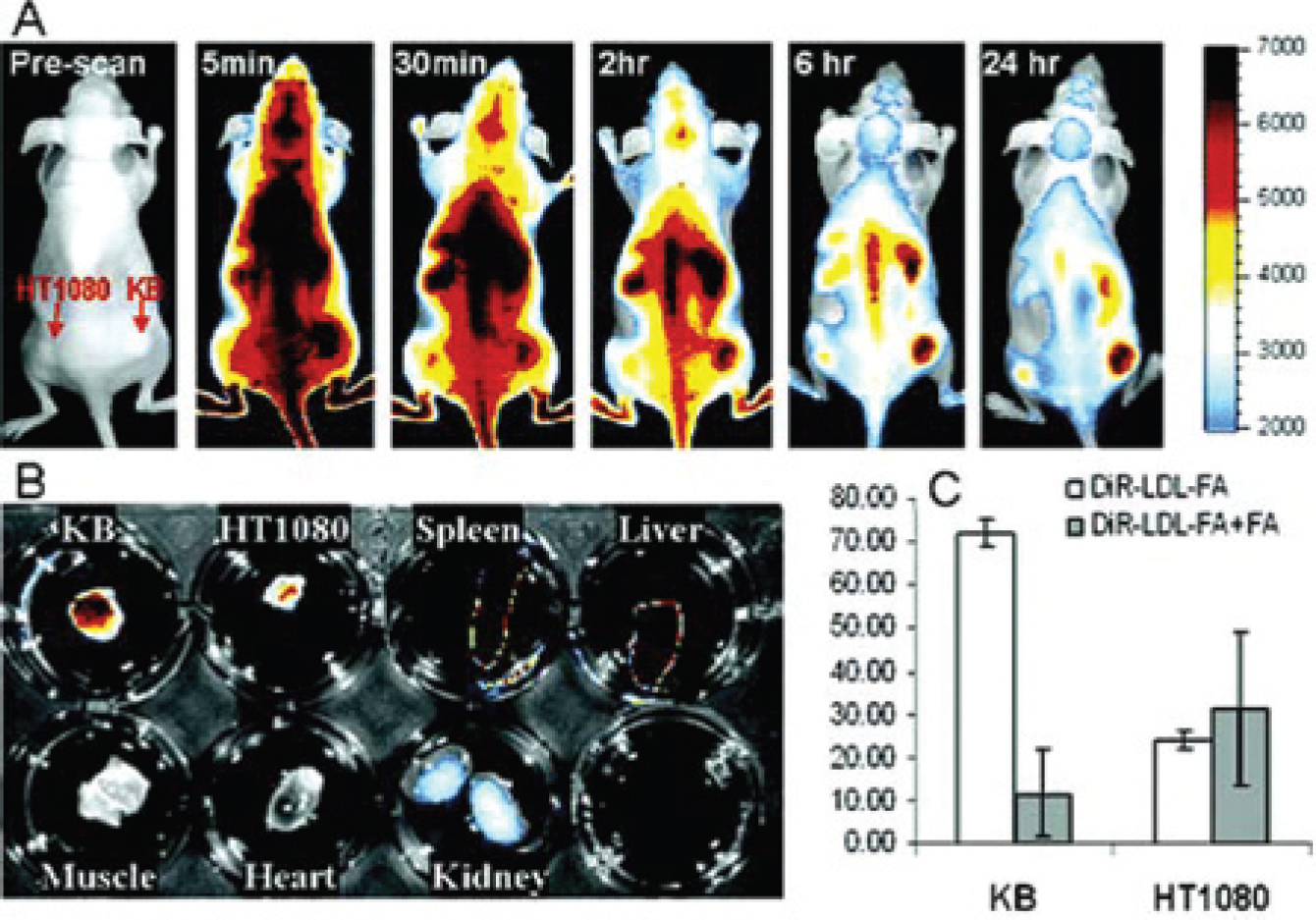
A, Real-time in vivo fluorescence images of KB/HT1080 dual tumor mice with intravenous injection of DiR–low-density lipoprotein (LDL)–folic acid (FA) (5.8 μM). B, Fluorescence images of tissues and tumors excised at 24 hours postinjection. C, Fluorescence readings of tumor extracts from in vivo FA inhibition assay. Figure reprinted from Chen J et al 26 by permission of The Journal of the American Chemical Society.
Other Lipoproteins
There is a whole family of lipoproteins spanning a wide range of dimensions; these include HDL (5–10 nm), LDL (20–25 nm), intermediate-density lipoprotein (3–40 nm), very-low-density lipoprotein (VLDL) (50–60 nm), and chylomicrons (> 1 μm). Appropriate labeling with receptor targeting or antigen targeting groups (eg, with antibodies or antibody fragments) and surface or core loading with NIRFs, MRI contrast agents, PET agents (eg, chelated 64Cu or 68Ga surface loaded on the phospholipid monolayer), or SPECT agents (covalently attached 123I or chelated 111In) enable use for diagnostic purposes. Alternatively, by loading these particles with drugs (see below) or radionuclides emitting high-energy gamma rays (eg, 67Cu), they can be used for targeted therapy. Some of these lipoproteins are internalized, whereas others are not. The pharmacokinetic properties of these agents will differ with their size; thus, HDL should prove superior to LDL for delivery to cell surface receptors but may also bind to receptors in the gastrointestinal tract, whereas the larger particles will show preferential binding to vascular targets. Studies have been initiated with HDL and VLDL by Zheng's laboratory. 27 Fayad's laboratory has pioneered the use of HDL for multimodality imaging of atherosclerotic plaque.28,29 The use of a surface-modified chylomicron remnant for delivery of computed tomography contrast agent has been reported. 30
Drug-Loaded LDL
Development of drug-delivering LDL has been initiated (W. Cao, A. Popov, J.D. Glickson, G. Zheng, unpublished study, 2006). The protein was alkylated with FA for delivery to FRs and core-loaded with a lipophilic conjugate of paclitaxel, as schematically indicated in Figure 8. DiI was surface-loaded for fluorescence monitoring. In vivo experiments are in progress with the KB tumor model (data not shown).
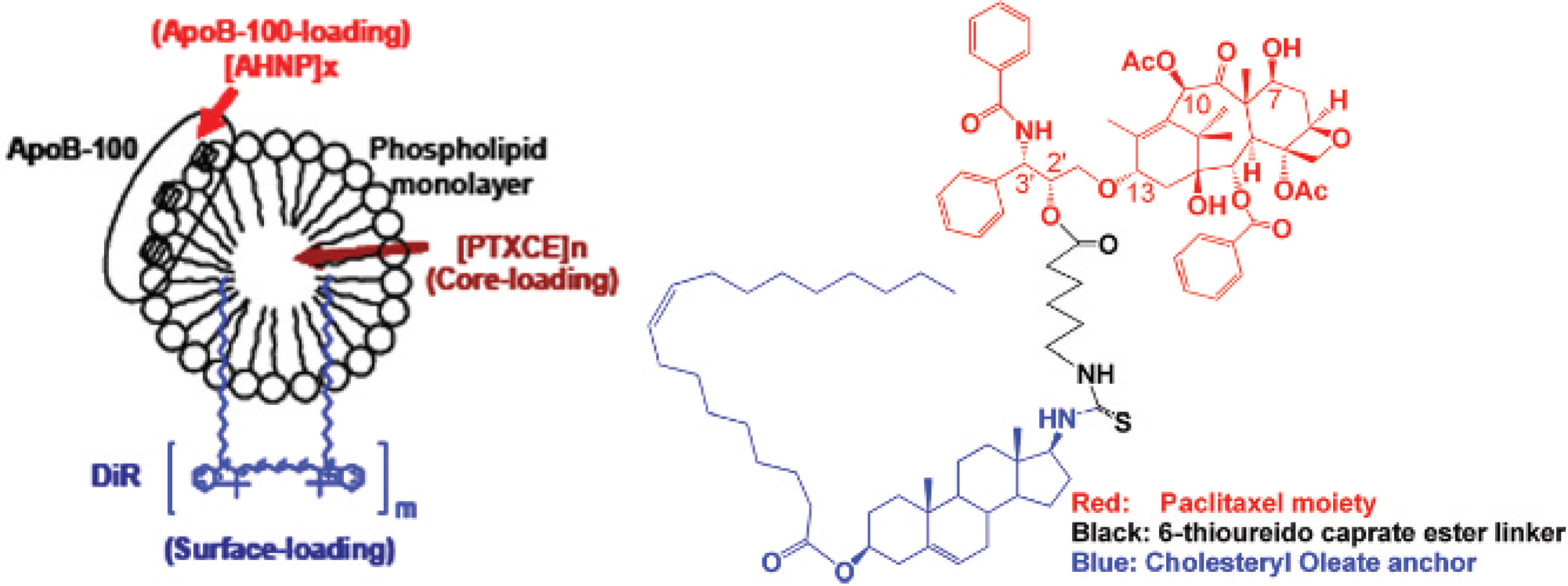
Schematic of lipoprotein-based nanoparticle protein loaded with folate and core-loaded with a lipophilic paclitaxel derivative. ApoB-100 = apolipoprotein B-100; PTXCE = paclitaxel cholesterol ester (oleate).
Superparamagnetic Iron Oxide Nanoparticles
Superparamagnetic iron oxide (SPIO) nanoparticles have been used as highly sensitive MRI markers for tracking cells and antibodies.31–35 Weissleder's laboratory attached dextran-coated SPIOs to transferrin and delivered these particles to tumors via transferrin-receptors.34,35 However, the tumors had to be transfected to prevent down-regulation of the transferrin receptors at high cellular iron concentrations. Given that lipoprotein receptors are not downregulated under these conditions, we have sought to core-load SPIOs into LDL. Similarly, iron oxide has been used for cell tracking, but the iron is diluted as the cells divide.31–33 By delivering iron in situ in the animal to cell surface receptors on the targeted cell, we should be able to overcome these limitations. Efforts to load LDL with iron oxide have proven unsuccessful even after lipid coating of the iron oxide particles (A. Stolpen and J.S. Leigh, personal communication, 1996). However, Choi and colleagues developed methods for targeting SPIOs to folate receptors 36 and for incorporating SPIOs into semisynthetic lipoproteins. The latter strategy is depicted in Figure 9. Using 5 nm SPIO clusters, lipoprotein–iron oxide particles with apolipoprotein A1 for targeting HDL receptors have been prepared; alternatively, apolipoprotein E can be used for targeting LDLR or synthetic 4F apolipoprotein A-1 mimetic amphipathic α-helical D-retropeptides. 37 These particles can also be redirected to folate or other receptors by alkylation of the lysine side chains. Such targeted particles can be used for T2-weighted MRI and for delivery of therapeutic agents to specific tumor receptors, vascular receptors on atherosclerotic plaque, stem cells, or macrophages.

Scheme for preparation of semisynthetic lipoprotein–iron oxide (IO) particles. The apolipoprotein can be A1 or E for targeting high-density or low-density lipoprotein receptors, respectively.
Advantages and Disadvantages of Lipoprotein-Based Nanoparticles
In summary, the lipoprotein-based lipoproteins constitute a highly versatile natural platform for delivery of diagnostic and therapeutic agents. In the absence of extensive chemical modification, they should be non-immunogenic, thereby avoiding a major potential limitation of synthetic nanoparticles. They are multifunctional and can be targeted at their natural receptors or modified to bind other natural receptors or perhaps even to unique unnatural receptors that could be transfected into cells (N. Sachs, personal communication, 2005). They provide a highly efficient mechanism for amplification of NIR, MRI, PET, or SPECT signal detection of the targeted cells in tumors, atherosclerotic plaque, or stem cells. Multivalency can be achieved by binding more than one targeting agent or therapeutic agent to the particles. In a similar manner, the platform facilitates multiple imaging modalities.
A key limitation is the existence of receptors on normal cells, such as the reticular endothelial system that leads to high background binding. This can be overcome to some extent by judicious choice of the targeting particle dimensions, by attaching polyethylene glycol groups to the surface to minimize nonspecific binding, or by choice of appropriate receptors, but it cannot yet be totally eliminated and remains a confounding problem in the use of this delivery system.
Many of the lipoproteins are isolated from human blood. Consequently, there is concern about avoiding introduction of pathogens that produce hepatitis or acquired immune deficiency syndrome (AIDS). Commercial sources are available for providing pathogen-free blood proteins, but this increases the cost of lipoproteins (CSL Behring-Biotherapies; http://www.cslbehring.com). Small proteins such as human apolipoproteins A1 and E can be produced in a recombinant form from bacterial sources, but the cost of these is quite high. Overall, commercialization of this technology is feasible, but the cost of the agents is likely to be high.
Footnotes
Acknowledgment
We are indebted to Dr. Theo van Berkel of Leiden University for providing the B16 melanoma and HepG2 hepatoma cell lines used in this study.