
Abstract
Adult neurogenesis is a highly dynamic process modulated by several pathologic and environmental factors, as well as by various compounds. So far, available techniques to study neurogenesis are lengthy and personnel and cost intensive. We developed a new tool based on the doublecortin promoter driving the expression of the luciferase reporter gene (DCX-promo-luciferase) in transgenic mice to perform in vivo imaging of neurogenesis. Indeed, the DCX-promo-luciferase mice allowed optical in vivo imaging of the onset of and increase in neurogenesis in developing fetal brains, as well as imaging of neurogenesis in the intact adult mouse central nervous system. Moreover, the capacity to specifically detect a small number of migrating neuronal precursors in vivo after transplantation is for the first time feasible using this DCX-promo-luciferase transgenic tool. The present imaging approach offers several crucial advantages over methods currently available, such as bromodeoxyuridine incorporation or labeling using iron oxide nanoparticles. Hence, it allows longitudinal study of neurogenesis in intact animals without the requirement of cellular prelabeling. Moreover, it guarantees that detection is specific for neuronal precursors and restricted to viable cells. Hence, our DCX-promo-luciferase transgenic model constitutes an effective tool that answers the pressing need for rapid investigation of the impact on neurogenesis of a large number of candidate compounds waiting to be tested.
INTEREST IN ADULT NEUROGENESIS has tremendously increased after its detection in the adult human hippocampus and olfactory bulb.1–3 Thus, the continuous generation of new neurons, which is confined to regions of adult neurogenesis, may play an important role in cognitive and emotional processes under physiologic and pathologic conditions. 4 Moreover, it could provide the basis for functional and cellular brain repair. To date, however, a simple tool for in vivo visualization and quantification of neurogenesis such as optical or bioluminescent imaging, magnetic resonance imaging (MRI), or positron emission tomography is lacking for rodents and humans. 5 Currently, the vast majority of studies addressing the extent and the kinetics of neurogenesis are still based on mitotic markers, such as bromodeoxyuridine (BrdU), which stably integrate into the newly synthesized deoxyribonucleic acid (DNA) of every proliferating cell. A posteriori, the fate of the labeled cells must be further analyzed with immunohistologic characterization to determine the phenotype and distribution of labeled cells. Unfortunately, this precludes the temporal analysis of neurogenesis in the same individual at different time points during the course of a disease or to monitor a therapeutic intervention.
Recently, we and others made use of paramagnetic markers, such as iron oxide nanoparticles, to detect in vivo and follow grafted neural stem cells using MRI. Cells to be grafted must first be loaded with these particles in vitro before they can be implanted in the brain and be detected by MRI as hypodense areas. 6 This technique provides very accurate morphologic information of the grafted cells localization over time and hence constituted a significant advance for the imaging of grafted cell in the central nervous system (CNS). This labeling strategy, however, has four major pitfalls. First, it cannot be determined if the detected nanoparticles are still within the grafted cells or have been released into the environment or even phagocytosed by surrounding cells. Second, the signal is independent of the cells' viability. Third, cells need to be labeled ex vivo; thus, this technique cannot be applied for analysis of endogenous neurogenesis. Finally, this labeling strategy is unspecific and thus provides no information about cell fate, such as cell integration, differentiation, and maturation of the grafted cells. Hence, there is an urgent need for improved methods to detect endogenous neurogenesis and grafted neural precursor cells.
An ideal marker for neurogenesis should thus be expressed transiently and specifically in neuronal precursors since this cell population reflects the rate of ongoing neurogenesis. 7 We previously demonstrated that doublecortin (DCX) was specifically and transiently expressed in the population of neuronal precursors and young neurons and thus constituted an ideal marker of neurogenesis.7–10 During developmental and adult neurogenesis, DCX was found to be expressed very early after neuronal determination of neural precursors, and anti-DCX antibodies could be used to efficiently detect newly generated neurons under physiologic conditions or those recruited by cerebral lesions, for example. In this regard, we showed that the presence and rate of neurogenesis could be quantified through the detection of DCX expression.7,10 Moreover, we identified the neuronal precursor-specific promoter region of the DCX gene 8 and used this element to drive a fluorescent reporter gene expression specifically in neuronal precursors and young neurons. 9 Here we describe a new transgenic tool designated DCX-promo-luciferase that enables in vivo optical imaging of developmental or adult endogenous neurogenesis and gives the possibility to follow the fate of transplanted neural progenitor cells.
Materials and Methods
Transgenic Mice
All experiments were carried out in accordance with the European Communities Council Directive of November 24, 1986 (86/609/EEC), and were approved by the local governmental commission for animal health. For generation of transgenic mouse lines, the vector phuDCX3509-FFLuci 8 was linearized with KpnI and BamHI. A fragment of 5.5 kb bearing the human DCX regulatory sequences (3.5 kb) and the reporter gene were purified and injected in B6C3F1 mouse embryos at one cell stage according to standard procedures (Transgenic Mouse Unit, Clinical Research Institute, Montreal, QC). Transgenic animals were identified by polymerase chain reaction (PCR) analysis for the presence of transgene in their genomic tail DNA using the following primers: FFLuciFow TCA AAG AGG CGA ACT GTG TG and FFLuciRev TTT TCC GTC ATC GTC TTT CC. Transgenic mice were further backcrossed in C57Bl6/J and thereafter designated DCX-promo-luciferase mice. DCX-promo-luciferase mice reproduced normally and did not show any overt morphologic or behavioral phenotypes. Moreover, CNS structures appeared to be normal at the microscopic level.
Cell Culture and Characterization
E14.5 embryos were collected from pregnant DCX-promo-luciferase heterozygous females. The telencephalon from each embryo was dissected and mechanically dissociated separately in six wells/plates as neural cell cultures. Genomic DNA was prepared from a remaining fragment of the embryos using the Wizard SV Genomic DNA Purification System (Promega, Mannheim, Germany) and used for PCR detection of the luciferase transgene. Cells from transgenic embryos were pooled together and further expanded in vitro.
Neural progenitor cells were grown in suspension and expanded in neurobasal medium (Gibco BRL, Invitrogen, Karlsruhe, Germany) supplemented with B27 (Gibco BRL), 2 mM L-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin, 2 μg/mL heparin, 20 ng/mL bFGF-2 (R&D Systems, Wiesbaden-Nordenstadt, Germany), and 20 ng/mL epidermal growth factor (EGF) (R&D Systems). For passaging, enzymatic dissociation was performed using Accutase (Innovative Cell Technologies Inc. San Diego, CA), as described previously. 11
Grafting (see below) was realized with proliferating neural progenitor cell cultures at passage 2. Prior to grafting, 1 μM of BrdU was added to the proliferation medium for a period of 24 hours. Immunohistologic characterization was realized on cultures between passages 3 and 5. To this end, neural progenitor cells were plated on polyornithin and laminin-coated coverslips at a density of 7.5 × 104 cells/cm2. Differentiation was induced by incubating for 5 days neural progenitor cells in growth factor-free medium supplemented with 1 μM of retinoic acid and 1% fetal calf serum.
For immunohistologic characterization of cell cultures, coverslips were fixed using a phosphate-buffered 4% paraformaldehyde solution pH 7.4 for 30 minutes. Blocking, antibody dilution, and washes were realized in fish skin gelatin buffer as described in Karl and colleagues. 8 The following primary antibodies were used: mouse antirat nestin (BD Biosciences Pharmingen, Erembodegem, Belgium, 1:500), goat anti-DCX C18 (Santa Cruz Biotechnology, Heidelberg, Germany, 1:500), rabbit anti-glial fibrillary acidic protein (GFAP) (Dako, Hamburg, Germany, 1:1000), mouse anti-βIII-tubulin (Promega, 1:500), and mouse anti-2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase) (Chemicon, Millipore, Illkirch, France, 1:200). Alexa Fluor-conjugated species-specific secondary antibodies were used (Invitrogen, 1:500). Nuclear counter-stain was obtained using DAPI (Sigma, Taufkirchen, Germany). Finally, coverslips were mounted in Prolong Gold Antifade Reagent (Invitrogen).
Surgical Procedures
Grafting procedures were performed in NMRI Nu/Nu nude mice. Two days prior to cell implantation, immunosuppression of mice was started and continued for the duration of the experiment via a daily intraperitoneal injection of cyclosporine (10 mg/kg of body weight). Before surgery, mice were deeply anesthetized through inhalation of 3% isoflurane in a mixture of 70% O2 and 30% NO2. Then, using a pulled-glass capillary and a Picospritzer II (General Valve, Parker Hannifin GmbH & Co. KG, Kaarst, Germany), 100,000 cells, contained in 2 μL artificial cerebrospinal fluid, were stereotactically delivered into the right ventricle (AP + 0.26; ML −0.8; DV −2.75 from the bregma according to the mouse brain in stereotaxic coordinates 12 ).
For the characterization of grafted cells' fate, mice were perfused with paraformaldehyde and tissue processed for histology as described in Couillard-Despres and colleagues. 9 The following primary antibodies were employed: goat anti-DCX C18 (Santa Cruz Biotechnology, 1:500), mouse anti-NeuN (Chemicon, 1:500), rabbit anti-GFAP (Dako, 1:500), mouse antinestin (BD Biosciences Pharmingen, 1:500), and rat anti-BrdU (Morphosys AbD GmbH, Dusseldorf, Germany, 1:250). Species-specific Alexa Fluor-conjugated secondary antibodies were used for detection (Invitrogen, 1:500). Finally, sections were mounted in Prolong Gold Antifade Reagent (Invitrogen) and analyzed on a confocal scanning laser microscope (Leica TCS-NT, Leica Microsystems GmbH, Wetzlar, Germany).
Imaging
In vitro luciferase activity detection was performed in white 96 wells/plates (Greiner Bio-One GmbH, Solingen, Germany). Neural cell cultures were dissociated into single cells using Accutase and were seeded in triplicate at various concentrations in polyornithin- (250 μg/mL) and laminin- (5 μg/mL) coated 96 wells/plates 4 hours before recording. Luciferase activity was detected using the Luciferase Assay System (Promega) and the Glomax Microplate Luminometer (Promega). In vivo detection of bioluminescence was performed using an IVIS Lumina (Xenogen, Caliperl Life Sciences GmbH, Russelsheim, Germany), except for imaging of pregnant mice, which was performed using an IVIS 3D Imaging System. Mice were maintained under light inhalation anesthesia using 2.5% isoflurane in oxygen. To obtain an improved morphologic resolution in adult DCX-promo-luciferase mice, hair covering the head was removed using depilatory cream. Luciferin (300 mg/kg of body weight) was injected intraperitoneally.
For the quantification of the luciferase activity in tissues, homogenates of olfactory bulbs and subventricular zones from mice of 7 days and 8 months of age (n = 3 each age) were performed. Tissues were dissected and homogenized in 500 μL of lysis buffer from the Luciferase Assay System (Promega). Luciferase activity in 20 μL of each homogenate was measured in white 96 wells/plate by adding 100 μL of luciferin-containing solution according to the Luciferase Assay System protocol (Promega), and bioluminescence recording was performed using an IVIS Lumina (Xenogen). Protein concentration of homogenates was determined using a Bradford assay (Sigma).
Results
Using transgenic mice expressing the firefly luciferase under the control of the human DCX promoter (DCX-promo-luciferase), we could visualize neurogenesis in the intact mouse CNS by optical bioluminescent imaging. During mouse fetal brain development, onset of neurogenesis occurs at approximately day 10.5. A clear bioluminescent signal originating from the ongoing developmental neurogenesis was detected in pregnant females on embryonic day 14.5 (Figure 1, A-C), and signal intensity increased further until birth, in accordance with the appearance and accumulation of newly generated neurons. However, owing to the depth of the embryos within the abdomen and embryo- and peristaltic-associated movements, poor morphologic resolution of the signal was achieved.

Optical imaging of the endogenous neurogenesis in doublecortin (DCX)-promo-luciferase mice. Pregnant female mouse bearing DCX-promo-luciferase fetuses recorded at (A) 11.5 days, (B) 14.5 days, and (C) 18.5 days postcoitum. The inset in B shows signal recorded from an extracted E14.5 DCX-promo-luciferase fetus. The intensity scale in C applies to A to C. D, Overlay of the bioluminescence signal and photograph of a 1-day-old DCX-promo-luciferase mouse; the inset shows signal emanating from the spinal cord using a lower threshold of detection. E, Photograph of a 2-month-old DCX-promo-luciferase mouse. Note that the skull has been shaved to improve the signal detection. F, Bioluminescence signal from the animal shown in E overlaid on the photograph; the inset shows a higher magnification, revealing stronger signal intensities over the olfactory bulbs. Color-coded scale minima and maxima are expressed in photon × s−1 × cm2–1 × sr−1. G, The bioluminescence originating from the ventricular and olfactory bulb regions was integrated in DCX-promo-luciferase mice as a function of age and expressed in photon/s−1. A strong signal reduction occurred in the early postnatal weeks and thereafter stabilized around 2 months of age and slowly decreased only in the adult transgenic mice (7 days, n = 3; 25 days, n = 1; 85 days, n = 4; 210 days, n = 2; 248 days, n = 4; 348 days, n = 1). H, The tissue luciferase activities generating the bioluminescence measured in G were directly quantified in olfactory bulb and subventrical zone (SVZ) homogenates from 7-day-old (n = 3) and 8-month-old (n = 3) DCX-promo-luciferase mice. Luciferase activity is expressed in photon/s−1/μg of protein−1.
On the other hand, areas bearing large amounts of young neurons in an isolated E14.5 embryo (see Figure 1B, inset) or a newborn transgenic pup (Figure 1D) could be nicely resolved. In the latter case, the intense signal was colocalized with the lateral ventricles, cortex, olfactory bulbs, and, to a lower extent, cerebellum. Significantly lower signal intensity could also be recorded from the spinal cord (see Figure 1D, inset).
In a 2-month-old transgenic mouse (Figure 1, E and F), the bioluminescence signal was roughly 100 times lower than that detected in newborn animals. This is mainly due to the progressive decrease in rates of neurogenesis occurring with aging, as well as due to an increase in light absorption by the adult head. Nevertheless, one can readily identify the lateral ventricles as being the major site of neurogenesis in the adult brain. Signal clusters could also be recorded from the olfactory bulbs (Figure 1F, inset). As for the newborn animals, the bioluminescence signal originating from the neurogenesis at the dentate gyrus could not be resolved by this transgenic animal model.
The bioluminescence signal emanating from the ventricular and olfactory bulb regions of DCX-promo-luciferase mice was plotted as a function of age. As shown in Figure 1G, following an initial fast decrease of bioluminescence in the first postnatal weeks, the bioluminescence intensity stabilized at 2 months of age and only slowly diminished thereafter with aging.
In addition, the luciferase activity within the tissues was also measured directly in olfactory bulb and subventricular zone (SVZ) homogenates from 7-day-old and 8-month-old DCX-promo-luciferase transgenic mice (Figure 1H). In agreement with the in vivo bioluminescence measurements, a strong reduction (approximately 10-fold) in the luciferase activity per microgram of proteins was assessed in 8-month-old DCX-promo-luciferase transgenic mice tissues compared with the activities measured in 7-day-old transgenic mice.
To follow the fate of specific neuronal precursors over time, we stereotactically injected neural progenitor cells into the right lateral ventricle of adult nude mice. These cells were derived from E14.5 DCX-promo-luciferase fetal forebrains that were shortly expanded under neural stem cell culture conditions. 11 Figure 2A shows the bioluminescence intensity recorded from various E14.5 DCX-promo-luciferase cell concentrations in vitro. We obtained a linear correlation between the bioluminescence signal and the cell number. Significant bioluminescence could already be detected with as little as 500 cells per well (see Figure 2A).
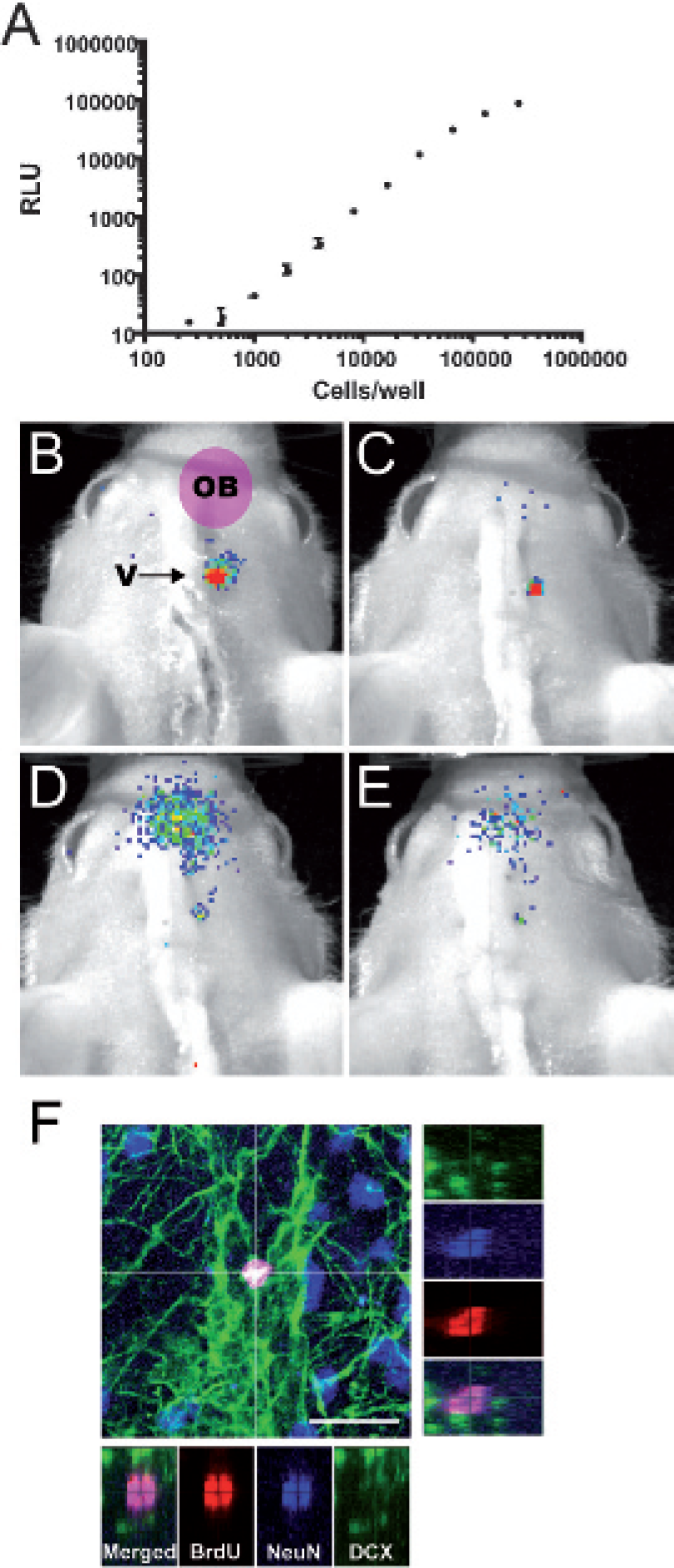
Optical imaging of doublecortin (DCX)-promo-luciferase-expressing neuronal precursors. A, A bioluminescence signal recorded from serial dilution of neural precursors in 96 wells/plate reveals a linear relationship between signal intensity and cell number. B to E, 105 cells were implanted in the right ventricle of NMRI Nu/Nu mice (arrow in B). Overlays of bioluminescence recordings and photographs show redistribution in a grafted mouse of emanating signals toward the olfactory bulb (OB) observed at (B) 6 days, (C) 8 days, (D) 10 days, and (E) 12 days postgrafting. The color-coded scale is identical from B to E. F, The fate of bromodeoxyuridine (BrdU)-labeled cells that migrated into the OB was investigated by immunohistology. The main panel shows a confocal micrograph stack in which a BrdU-labeled cell located at the proximal end of the OB can be seen. In this cell, BrdU (red) colocalized with the mature neuronal marker NeuN (NeuN in blue, colocalization with red appears as violet), but not with the immature neuronal marker DCX (green). The lateral small panels show as a cross section of the stack in the z-axis each confocal channel individually and merged, confirming colocalization of BrdU with NeuN. Scale bar represents 20 μm.
These neural progenitor cells were previously shown to be able to induce expression of the endogenous DCX gene and were able to activate DCX promoter constructs following differentiation. 8 Immunohistologic characterization under proliferating conditions of the neural progenitor cells used in this study revealed that virtually all of them were expressing nestin (98.5%), a marker used to detect putative neural stem cells. On the other side, DCX (young neuron marker) and GFAP (astrocyte marker) could be detected in only 0.7% and 0% of the cells, respectively. The expression of βIII-tubulin, another early neuronal marker that appears in vitro slightly prior to DCX, was detected in 2.3% of the cells, whereas CNPase (oligodendrocyte marker) was not detected. Following an incubation of 5 days in differentiation conditions, a strong decrease in the number of nestin-expressing cells could be observed as only 18.4% of the cells were still expressing nestin. In contrast, an increase was noticed for differentiation-specific markers, with 8.6% of the cells expressing βIII-tubulin, 3.6% of the cells expressing high levels of DCX, and 10.6% expressing GFAP. No cells were found to expressed CNPase following this differentiation protocol. The percentages of DCX-expressing cells were expected to increase with longer differentiation treatment since a large proportion of differentiating cells expressed low levels of DCX at this time point. Together, this indicates that our grafted proliferating neural progenitors were undifferentiated but were able to respond to differentiation cues in their environment.
Cells from the proliferating neural progenitor cultures were labeled with BrdU and then used as grafting material in nude mice. Dissociated cells (105 cells in 2 μL of artificial cerebrospinal fluid) were stereotactically injected into the right lateral ventricle. Once implanted, neuronal precursors adopted a similar behavior as endogenous precursors and migrated toward the olfactory bulb (Figure 2, B to E). Eight days following grafting, a bioluminescence signal could be clearly detected outside the graft site and was registered at the position of the olfactory bulb, revealing the expected colonization of this structure by grafted neuronal precursors. The bioluminescence intensity emanating from the olfactory bulb reached a maximum at day 10 after implantation and then began to regress. The recorded bioluminescent signals are hence in accordance with reported kinetics of migration and maturation of newly generated neurons in mice and rats.9,10
To monitor the fate of the grafted neural progenitor cells, mice were perfused and the phenotype of BrdU-labeled cells was investigated at 7 days and 28 days postimplantation. Immunohistologic analysis revealed that BrdU-labeled cells migrated to the olfactory bulb and were associated with the expression of DCX and NeuN (Figure 2F). Although glial differentiation or lack of differentiation of the grafted cells cannot be ruled out, no BrdU-labeled cells expressing nestin or GFAP could be detected in the olfactory bulb (data not shown). Hence, in vivo bioluminescence signals detected in grafted mice were truly generated by a population of neural progenitor cells that differentiated into young neurons migrating and integrating in the olfactory bulb.
Discussion
In summary, we present a new mammalian animal model that is to date the first to allow optical in vivo imaging of neurogenesis and to assess the neuronal fate of transplanted progenitors. The use of the DCX promoter to drive the expression of the firefly luciferase reporter gene in transgenic animals enables us to detect endogenous, developmental, and adult neurogenesis and to monitor a specific population of grafted neuronal precursors.
The capacity to follow neurogenesis and changes in neurogenesis over time in an individual animal constitutes a major advantage of the DCX-promo-luciferase model we developed over other conventional methods to analyze neurogenesis, such as BrdU labeling of proliferating cells. Since no ex vivo prelabeling of cells is required, the DCX-promo-luciferase transgenic model allows the analysis of changes in endogenous neurogenesis occurring, for example, during development or aging. Therefore, DCX-promo-luciferase mice now provide a means to investigate the physiologic role of neurogenesis. Moreover, this mouse system might provide a tool to monitor the impact of CNS lesions on neurogenesis, for example, the induction of neurogenesis in stroke models or during neurodegenerative processes13,14 over time.
The increasing number of genetic mouse models for neurodevelopmental and neurodegenerative diseases further fosters the need for neurogenesis reporter models suitable for in vivo imaging. Our DCX-promo-luciferase tool thus constitutes a new means to monitor neurogenesis in vivo along the course of the disease in these various mouse models or following therapeutic intervention. In vivo imaging of neurogenesis using the DCX-promo-luciferase mice will also help identify and validate drug candidates for therapy of neurologic and psychiatric diseases that are likely associated with neurogenesis. Owing to the time-, labor-, and cost-intensive conventional methods to analyze neurogenesis, 15 only a limited number of such active molecules have been identified using candidate approaches. 16 Systematic screenings of compound libraries acting on neurogenesis and validation by in vivo imaging of neurogenesis have not been performed so far, but the present work offers an urgently needed test system.
Bioluminescence is currently the best-suited technology for optical imaging of deep brain structures in animal models. In addition, a promoter-based bioluminescence strategy offers the key advantage that only viable cells will be detected since the light emission of the firefly luciferase is adenosine triphosphate dependent. Moreover, the transgene cannot be transferred to surrounding cells and the DCX promoter is selectively active in neuronal precursors and young neurons. This ensures the specific identity of the cell population detected.
Our DCX-promo-luciferase transgenic tool allows us to monitor neuronal cell fate in vivo without the requirement of in vitro prelabeling. This is in contrast to the present nanoparticle-based strategies employed to image neural stem cells in the CNS by MRI.6,17 Although detection of iron oxide nanoparticle-loaded cells by MRI offers a very good morphologic resolution, this approach presents disadvantages: cells under investigation need to be prelabeled in vitro prior to implantation in the CNS. These steps open the door to possible toxicity or transformation of the cell identity owing to in vitro conditions and from the labeling procedure per se. Also, if labeled cells proliferate, they will gradually dilute their content of nanoparticles and will become undetectable. Another problem intrinsic to the nanoparticle approach is that their MRI signal is dependent on neither the cell type nor on the cell viability. Thus, it is not possible using nanoparticles to assess the survival of grafted cells or their fate in terms of differentiation. Moreover, surrounding host cells can potentially phagocytose released nanoparticles, thereby creating confounding results.
Adult neurogenesis is a highly dynamic process that may have a physiologic role in cognitive and emotional functions and could be associated with disorders such as schizophrenia and depression.4,18 Moreover, neurogenesis is modulated by several pathologic and environmental factors; for a review, see Ming and Song. 19 To date, mechanisms by which pathologies or various compounds influence neurogenesis are yet to be unraveled. Nevertheless, it is tempting to see a link between the deficient olfactory perception documented in the vast majority of Parkinson disease patients and the altered olfactory bulb neurogenesis we reported in animal models of Parkinson disease.20–22 Modulation of neurogenesis might hence be a crucial element of future therapeutic strategies in cases of neurodegeneration and cerebral lesions. However, investigation of such issues is currently limited by the lack of efficient in vivo screening and validation tools allowing effective translational studies. Therefore, our DCX-promo-luciferase model for in vivo imaging of neurogenesis provides an answer to a pressing need and offers an attractive system for monitoring neurogenesis under normal physiologic and under various pathophysiologic situations.
Footnotes
Acknowledgments
We are thankful to Xenogen Corp. (Alameda, CA) for generous use of the IVIS 3D Imaging System and to Prof. Edinger, funded by DFG KFO146, for providing access to the IVIS Lumina.