
Abstract
Surgical resection remains a definitive treatment for prostate cancer. Yet, prostate cancer surgery is performed without image guidance for tumor margin, extension beyond the capsule and lymph node positivity, and without verification of other occult metastases in the surgical field. Recently, several imaging systems have been described that exploit near-infrared (NIR) fluorescent light for sensitive, real-time detection of disease pathology intraoperatively. In this study, we describe a high-affinity (9 nM), single nucleophile-containing, small molecule specific for the active site of the enzyme PSMA. We demonstrate production of a tetra-sulfonated heptamethine indocyanine NIR fluorescent derivative of this molecule using a high-yield LC/MS purification strategy. Interestingly, NIR fluorophore conjugation improves affinity over 20-fold, and we provide mechanistic insight into this observation. We describe the preparative production of enzymatically active PSMA using a baculovirus expression system and an adenovirus that co-expresses PSMA and GFP. We demonstrate sensitive and specific in vitro imaging of endogenous and ectopically expressed PSMA in human cells and in vivo imaging of xenograft tumors. We also discuss chemical strategies for improving performance even further. Taken together, this study describes nearly complete preclinical development of an optically based small-molecule contrast agent for image-guided surgery.
Keywords
Introduction
In 2005, prostate cancer will be diagnosed in approximately 232,090 men in the United States, and 30,350 previously diagnosed men will die of the disease [1]. With the development of PSA and other screening markers, a majority of men will be diagnosed with early-stage disease, and many men will be candidates for surgical resection. Despite a debate in the literature about optimal therapeutic strategy, most well-selected patients will be cured by surgery. Surprisingly, prostate and other cancer surgeries are still performed “blindly,” without any type of intraoperative image guidance. Prostate cancer, in particular, would benefit from intraoperative imaging of the prostate boundary, of prostate cancer extension through the capsule, of in-transit metastases, and of frank metastases in pelvic lymph nodes. Such imaging will require targeted delivery of contrast agents.
Of the many molecular targets that have been described for the prostate gland and prostate cancer, probably none has received more attention than PSMA. PSMA is a 100-kDa type II transmembrane glycoprotein with a restricted pattern of tissue expression. It is highly expressed on normal and malignant prostate epithelial cells and was originally cloned [2] as a LNCaP cell-surface antigen of unknown function. PSMA is also expressed on the surface of neurons, where it was independently cloned as the major N-acetylated α-linked acidic dipeptidase (NAALADase or glutamate carboxypeptidase II [3]). PSMA was independently cloned a third time from pig jejunum [4] as the major lumenal pteroyl-poly-γ-glutamate carboxypeptidase (folate hydrolase) activity. Cell-surface PSMA is internalized constitutively as well as through antibody-induced endocytosis [5], and can be shed under certain conditions [6].
Many properties of PSMA suggest that it may be an ideal target for the detection and treatment of human prostate cancer. Most importantly, in the systemic circulation, it is mainly restricted to benign and malignant prostatic epithelial cells, where it is abundant. As prostate cancer evolves to a hormone refractory state, both levels of PSMA and the ratio of the major (transmembrane) splice variant to the minor (cytosolic) splice variant, greatly increase [7]. Surprisingly, PSMA is also expressed on the neovascular endothelium of most human solid tumors, suggesting that it plays an essential function in the development and/or progression of prostate and other cancers [8,9].
Molecules that target PSMA have been the subject of intense research. To date, PSMA-specific antibodies [8–10], aptamers [11], peptides [12], peptide derivatives [13], and small molecules [14,15] have been described. From these have emerged radiolabeled antibodies for imaging [16], radiolabeled antibodies for therapy [17, 18], drug-filled nanoparticles [19], thrombosis-inducing molecules [13], and targeted chemotherapeutics [20]. Low-molecular-weight ligands, and especially small molecules, are highly preferred for tumor targeting due to their rapid biodistribution, rapid clearance, improved tumor penetration, and ease of synthesis.
Over the last 3 years, there has been significant interest in using near-infrared (NIR: 700-900 nm) fluorescent light for in vivo imaging (reviewed in Ref. [21]), and specifically intraoperative imaging. Indeed, three intraoperative NIR fluorescence imaging systems are currently under development: the Spy™ system from Novadaq (Mississauga, Ontario, Canada), the Palomar™ system from Spectros (Portola Valley, CA), and a simultaneous color video/NIR fluorescence system developed by our laboratory and licensed to GE Healthcare [22]. Our laboratory has also developed robust methods for creating small molecule-[23], peptide-[23], and protein-based [24] contrast agents using tetra-sulfonated heptamethine indocyanine NIR fluorophores.
To date, a NIR fluorescent small molecule targeted to PSMA has not been described. In this study, we report the synthesis and purification of a novel NIR fluorescent contrast agent specific for PSMA. We also demonstrate comprehensive characterization of this targeted optical contrast agent in vitro and in vivo.
Materials and Methods
Reagents
The NHS ester of IRDye78 (IRDye78-NHS) was purchased from LI-COR (Lincoln, NE). The carboxylic acid form (IRDye78-CA) was kindly provided by LI-COR. ICG and IR-786 were purchased from Sigma (St. Louis, MO). All fluorophores were stored as dry powders under nitrogen at −80°C until use. Guilford 11254-36 (GPI (2-[((3-amino-3-carboxypropyl)(hydroxy)phosphinyl)-methyl]pentane-1,5-dioic acid) was synthesized according to the previously published procedure [25]. β-
Synthesis and Purification of NIR Fluorescent Small Molecules
All procedures were performed under reduced light conditions. Synthesis and purification of β-AG-78 has been described previously [23]. Covalent conjugation of IRDye78 to GPI (GPI-78) was performed by the addition of 0.1 mL of 300 mM triethylamine in ultradry DMSO to 0.2 mL of a 10 mM solution of GPI in ultradry DMF. After 5 min, 0.2 mL of 15 mM IRDye78-NHS in ultra-dry DMSO and 0.5 mL of ultra-dry DMSO were added to the reaction mixture. Constant stirring in the dark was maintained overnight at room temperature (RT). Preparative scale GPI-78 was purified by reverse-phase HPLC using a Waters (Milford, MA) chromatography system equipped with a Symmetry Prep™ C18 column (19 × 150 mm, 7 µm particle size) and a dual-wavelength absorbance detector (set to 254 and 700 nm). Using a flow splitter (Upchurch Scientific, Oak Harbor, WA), a portion of the eluate flowed into a Sedex model 75 (Richards Scientific, Novato, CA) evaporative light scatter detector with the nebulizer modified to reduce band broadening at low flow rates. Solvent A was 10 mM TEAA and Solvent B was absolute methanol. The purification utilized a linear gradient from 15% to 50% Solvent B in 30 min, beginning 10 min after injection. Mobile phase flow rate was 15 mL/min. GPI-78, eluting with a retention time of 21 min from the start of the gradient, was collected and concentrated on an Oasis HLB desalting cartridge (Waters).
ES-TOF Mass Spectroscopic Analysis
The purities of all compounds used in this study were measured using LC/MS on a Waters HPLC system equipped with a Micromass (Waters) LCT ES-TOF mass spectrometer. Analysis of GPI-78 was performed on a Symmetry (Waters) C18 column (4.6 × 150 mm, 5 µm particle size) with the same gradient used for purification except it was over 25 min, starting at 3 min, and the flow rate was 1 mL/min. ES-TOF parameters included negative mode, capillary voltage of −2700 V, extraction cone voltage of −3 V, and a sample cone voltage of −50 V.
Characterization of Photoproperties
Absorbance spectrometry was performed in a 1 cm path length quartz cuvette (Starna, Atascadero, CA), mounted in a CUV-ALL-UV four-way cuvette holder (Ocean Optics, Dunedin, FL), and excited with a balanced deuterium-tungsten light source (Ocean Optics). Absorbance measurements were made on a USB2000 (Ocean Optics) spectrometer with a 1 nm resolution from 200 to 870 nm, using 5 µM of NIR fluorophore in the indicated buffer. Fluorescence spectrometry was performed in a three-sided quartz cuvette (Starna) excited with a 5 mW 670 nm laser diode coupled through a 300 µm core diameter, NA 0.22 fiber (Fiberguide Industries, Stirling, NJ). Fluorescence measurements were made on an HR2000 (Ocean Optics) spectrometer with a 6.7 nm resolution from 200 to 1100 nm, using 1 µM of NIR fluorophore in the indicated buffer. Data acquisition was performed on a Dell computer using the OOIBase32 software (Ocean Optics). Quantum-yield (QY) measurements were performed using ICG in DMSO (QY 13% [26]) as calibration standard, under conditions of matched fluorophore absorbance.
Expression and Purification of the Enzymatically Active Extracellular Domain of PSMA from Insect Cells
Plasmid PSMA2, containing the full-length cDNA of human PSMA, was the kind gift of Dr. Joseph T. Coyle (McLean Hospital, Belmont, MA). The extracellular domain (amino acids 44–750) was PCR-amplified from PSMA2 to generate flanking BamHI (5′) and NotI (3′) restriction sites using the following primers:
5′ Primer: 5′-ACGTACGTAGGATCCAAATCCTCCAATGAAGCTACT-3′
3′ Primer: 5′-ATCGATCGAGCGGCCGCTCAGGCTACTTCACTCAAAGTCTC-3′
The DNA product was gel-purified, digested with BamHI and NotI, re-gel-purified, and cloned into similarly digested vector pAcGP67BHis. Baculovirus expression vector pAcGP67BHis, containing an N-terminal histidine (His) tag, was the kind gift of Dr. Cary H.C. Lai (Scripps Research Institute, La Jolla, CA). All products were confirmed by DNA sequencing. The 81 kDa secreted His-PSMA protein has the sequence GSHHHHHHGGG preceding the wild-type PSMA extracellular domain sequence.
High-titer His-PSMA baculovirus was produced in Sf9 cells using BaculoGold™ linearized DNA (Pharmingen, San Jose, CA). Sf9 cells were grown in Grace's medium (Gibco-BRL, Gaithersburg, MD). High Five™ insect cells (Invitrogen, Carlsbad, CA) grown in Ex-Cell 400 (JRH Biosciences, Lenexa, KS) culture medium in spinner flasks were infected with His-PSMA baculovirus and supernatants were collected at 56 hr. Fifteen milliliters of supernatant was applied to each milliliter of packed Probond***™ (Invitrogen) nickel agarose beads. After rocking for 1 hr at RT, beads were washed five times with PBS, resuspended as a 50% suspension in PBS supplemented with 10% glycerol, flash frozen in liquid nitrogen, and stored at −80°C until use. His-PSMA was eluted from nickel agarose by resuspending the beads in two packed volumes of 250 mM imidazole in PBS, pH 7.4, vortexing briefly, rocking for 5 min at RT, and passing once through a 0.8 µm vacuum filter, then through a 0.2 µm vacuum filter. His-PSMA was buffer-exchanged into 50 mM Tris, pH 7.4 supplemented with 150 mM NaCl (TBS), and concentrated, using a 10 kDa Ultrafree-15 (Millipore, Bedford, MA) centrifugal filter at 2500 × g.
PSMA (NAALADase) Enzyme Assay
NAALADase enzymatic activity was quantified by measuring the hydrolysis of 3H-NAAG radiolabeled at the glutamate residue essentially as described by Robinson et al. [27]. Assays were performed in 96-well V-shaped polypropylene plates (Fisher Scientific) containing 50 mM Tris-HCl, pH 7.4, 30 nM 3H-NAAG, and 50 ng of His-PSMA protein in a total volume of 50 µL. Assays were initiated by the addition of His-PSMA, with or without inhibitors, and the plates incubated at 37°C for 20 min by floating in a water bath. Assays were terminated by addition of 50 µL of 100 mM phosphate buffer, pH 7.4. Monstr-pette™ pipettes (4.5 mL; Chase Scientific Glass, Rockwood, TN) were placed in a custom manifold and to each was added a 3-mm KG-33 borosilicate glass bead (Kimble, Vineland, NJ) and 1 mL of 200–400 mesh AG 1-X8 anion exchange resin with formate as the counterion (Bio-Rad, Hercules, CA). Aliquots (75 µL) of the quenched assay were applied to the resin. The cleaved 3H-glutamate was eluted by applying 2 mL of 1 M formic acid to each pipette and collecting eluant directly into scintillation vials. After the addition of 10 mL of Packard (Boston, MA) Emulsifier-Safe™ scintillation liquid (16.7% final aqueous/scintillant ratio) in each vial and vigorous shaking, radioactivity was measured on a Packard 2300TR scintillation counter. All experiments were performed independently at least three times, and all assays were performed in triplicate for each data point. Results were analyzed using a nonlinear curve-fitting program (sigmoidal dose response) from Prism (GraphPad Software, San Diego, CA).
For tumor NAALADase activity, tumors were excised at 250 mm3, diluted to 0.25 g per 0.25 mL of H2O, minced with scissors, homogenized, and sonicated on ice for 2 × 30 sec. Membrane and supernatant fractions were prepared by centrifugation at 50,000 × g. The membrane fraction was resuspended in 50 mM Tris, pH 7.5, and the supernatant fraction was similarly adjusted using 1 M Tris, pH 7.5.
Measurement of Affinity by Fluorescence Polarization
Assays were performed using a Beacon 2000 extended red fluorescence polarization system (PanVera, Madison, WI) equipped with a halogen lamp, a 750 ± 50 nm excitation filter, and a custom 800 nm emission filter. Temperature was regulated at 25°C. For saturation experiments, fixed concentrations of NIR fluorescent molecule were used (1 nM final concentration) and increasing concentrations of His-PSMA were added (the absolute concentration of active sites of PSMA was determined by 2-PMPA titration, data not shown). Polarization was performed in sterile-filtered 50 nM Tris-HCl, pH 7.4.
Cell Lines
For in vitro experiments, human prostate cancer cell lines LNCaP and PC-3 were obtained from the ATCC (Manassas, VA). Cell lines were cultured at 37°C under humidified 5% CO2 in RPMI 1640 medium (Mediatech Cellgro, Herndon, VA) supplemented with 10% fetal bovine serum (Gemini Bio-Products, Woodland, CA) and 5% penicillin/streptomycin (Cambrex Bioscience, Walkersville, MD).
Quantitative Cell Binding Assay
To calculate the number of cell-surface receptor sites occupied by GPI-78, LNCaP cells were incubated with 2 µM GPI-78 in TBS or PBS for 20 min at 4°C. After three washes with the same buffer, cells were lysed with 10% sodium dodecylsulfate (SDS) in PBS and the fluorescence of cell lysates was measured against a standard curve formed with similar cell lysates adjusted to known concentrations of IRDye78-CA. PSMA-specific net fluorescence was calculated by subtracting moles of fluorophore bound in TBS from that bound in PBS.
Ectopic Expression of Full-length Human PSMA and Erb-B2 Using Adenovirus
The full-length human PSMA cDNA from PSMA2 was shuttled through the custom vector pC, excised using BglII and NotI, gel-purified, and cloned directionally into similarly digested vector pAdTrack-CMV [28]. pAdTrack-CMV is an intermediate during adenovirus production that permits co-expression of a protein of interest and GFP under the control of separate CMV promoters. The full-length cDNA for human Erb-B2 (a kind gift from Dr. Kermit Carraway, UC Davis, Davis, CA) was excised using HindIII, gel-purified, and cloned into similarly digested pAdTrack-CMV. High-titer adenoviruses co-expressing GFP and either human PSMA or human Erb-B2 were produced after optimization of a previously published pAdEasy-1 protocol [28]. A detailed protocol is available from the authors.
In Vitro Fluorescence Studies
For adenovirus expression studies, exponentially growing PC-3 cells at a confluence of 50% on glass coverslips were infected at an MOI of approximately 10 with GFP/PSMA or GFP/Erb-B2 adenovirus, and assayed 48 hr later. Cells grown on coverslips were incubated with 0.2 mL of TBS containing 2 µM of GPI-78 for 15 min at 37°C. Cells were washed three times with TBS and fixed with 2% paraformaldehyde in TBS for 15 min at RT. The cells were then permeabilized with TBS supplemented with 0.1% Tween-20 (TBS-T), stained with mouse anti-PSMA monoclonal antibody 4D4 for 45 min in 0.5% nonfat dry milk/TBS-T then with Cy3-conjugated donkey anti-mouse secondary antibody (Jackson ImmunoResearch, West Grove, PA). After counterstaining with DAPI, coverslips were mounted using Fluoromount-G and imaged on a previously described four-channel NIR fluorescence microscope. For fluorescence studies of endogenous PSMA, the same protocol was employed without antibody staining using the cell lines and conditions specified above.
Blood Half-life Measurements
CD-1 mice (40 g) were injected intravenously with 10 nmol (0.25 µmol/kg) IRDye78-CA or GPI-78. At the time points indicated, tail blood was collected in a micro-hematocrit tube (Fisher Scientific) and NIR fluorescence measured against IRDye78-CA standards (0-2 µM) diluted in mouse blood supplemented with 5 mM EDTA.
Xenograft Tumor Models and In Vivo Imaging
Mice were used in accordance with an approved institutional protocol. Eight-week-old male hairless athymic NCI nu/nu mice were purchased from Harlan Sprague Dawley (Indianapolis, IN) and anesthetized with 50 mg/kg intraperitoneal pentobarbital. Subcutaneously on the right flank were injected 5 × 106 PSMA-positive LNCaP-C42 cells (Viromed Laboratories, Minnetonka, MN) resuspended in Matrigel (BD Bioscience, Bedford, MA). In approximately 2 weeks, LNCaP tumors were visible, and on the left flank were injected 5 × 106 of PSMA-negative human bladder cancer TsuPR1 cells resuspended in Matrigel. The latter were a generous gift of Dr. John T. Isaacs (Johns Hopkins University, Baltimore, MD). Within 15 days after implantation of TsuPR1 cells, each mouse developed bilateral tumors of approximately 5 mm in diameter.
For NIR fluorescence imaging, mice were anesthetized and injected intravenously with 5 nmol (0.2 µmol/kg) GPI-78 or IRDye78-CA diluted in TBS. Continuous imaging was performed on a custom small-animal NIR fluorescence imaging system. The system has been described in detail previously [22]. Briefly, it is composed of two wavelength-isolated excitation sources, one generating 0.5 mW/cm2 400–700 nm “white” light, and the other generating up to 50 mW/cm2 725–775 nm light over an 8-cm diameter field of view. Simultaneous photon collection of color video and NIR fluorescence images is achieved with custom-designed optics that maintain separation of the white light and NIR fluorescence (>795 nm) channels. After computer-controlled (LabVIEW) camera acquisition via custom LabVIEW (National Instruments, Austin, TX) software, anatomic (white light) and functional (NIR fluorescent light) images can be displayed separately and merged. All images are refreshed up to 15 times per second. The entire apparatus is suspended on an articulated arm over the surgical field, thus permitting noninvasive and nonintrusive imaging.
Results
PSMA Active Site Binding Molecules
In the original description of the NAALADase activity of PSMA, it was mentioned that the N-acetyl group could be removed from NAAG (Figure 1A) with only a 4–5-fold change in enzyme kinetics [27,29]. This suggested to us that the N-acetyl group did not contribute significantly to binding the substrate pocket of PSMA and that, possibly, the α nitrogen could tolerate bulk substitution with contrast agents. β-NAAG [29] (Figure 1A) is an N-acetylated competitive inhibitor of PSMA with micromolar affinity. By synthesizing the de-acetylated derivative of this molecule, β-AG (Figure 1A), we reasoned that a single nucleophile-containing “modular” ligand would be produced, with the α nitrogen available for convenient conjugation. Indeed, this exact molecule has been shown to direct targeting of intravenously injected therapeutics [13].

Synthesis, purification, and optical characterization of GPI-78. (A) Chemical structures and molecular weights of PSMA/NAALADase substrates, inhibitors, and ligands (top row), NIR fluorophores (middle row), and NIR fluorescent PSMA ligands (bottom row) employed in this study. (B) The primary amine of GPI was conjugated to the sodium salt of IRDye78-NHS in one step as described in Materials and Methods. (C) Purification of GPI-78. A combination of ELSD detection, absorbance detection, and mass spectrometry is able to identify all chemical reactants and products as they elute from the C18 column. The HPLC gradient is shown at the top. Mobile phase buffers were A = 10 mM TEAA, pH 7.0, and B = methanol. Detectors included absorbance at 700 nm (Abs700 nm), ELSD, and mass spectrometer ion current for 1309 Da species (IC1309 Da). Slight differences in retention time are due to finite tubing lengths among the three detectors. Ionization and salts of GPI-78 seen during ES-TOF analysis (bottom) are indicated. (D) Photoproperties of IRDye78 and GPI-78. Absorbance (left) and fluorescence (right) spectrometry was performed in PBS (top) and FBS (bottom). Also shown is a summary of optical properties of the parent NIR fluorophore IRDye78-CA and its GPI-78 conjugate.
After synthesis and testing of β-AG and β-AG-78 (see below; Figure 1A), it was obvious that higher affinity ligands would be necessary for optimal targeting. Based on the known potency of phosphinic acid inhibitors of PSMA such as 2-PMPA [30], and in collaboration with Guilford Pharmaceuticals, we explored the use of the single nucleophile-containing GPI compound (Figure 1A) and its derivatives as modular ligands for PSMA targeting.
Synthesis and Purification of GPI-78
GPI was conjugated covalently to IRDye78 (Figure 1B) to create the molecule GPI-78 using the strategy detailed in Materials and Methods. A reverse-phase HPLC purification procedure (Figure 1C) was developed to purify GPI-78 in preparative quantities. From 0.6 mg of GPI starting material, the typical final yield of GPI-78 was 1.6 mg (60%). Purity of the final product measured using analytical LC/MS was −95%, and ES-TOF mass spectrometry confirmed the expected masses (Figure 1C).
GPI-78 Photoproperties in PBS and Serum
IRDye78 was chosen as the NIR fluorophore for contrast agent development because of its excellent chemical and optical properties (Figure 1A and D). In particular, tetra-sulfonation renders the molecule soluble at concentrations of at least 10 mM in aqueous buffer and contributes to the relatively high QY of 9%. Tetra-sulfonation also minimizes the interaction of fluorophore with serum proteins, as evidenced by the insensitivity of IRDye78-CA and GPI-78 to 100% FBS (Figure 1D). Peak absorbance of GPI-78 in PBS and FBS was 773 and 778 nm, respectively. Unlike protein conjugation, which often leads to moderate bathochromatic shifts [24], conjugation of IRDye78 to GPI resulted in no significant change in peak excitation, peak emission, or QY (Figure 1D).
Expression of Enzymatically Active Recombinant PSMA in Insect Cells
The entire extracellular domain of PSMA (amino acids 44–750; Figure 2A), which contains the active enzyme, was expressed as a secreted protein in insect cells. This molecule is longer than a baculovirus-produced extracellular PSMA described previously [11], but does not contain any intracellular or membrane-spanning domain [31]. An engineered N-terminal His tag permitted convenient purification of His-PSMA on nickel agarose (Figure 2B). Final yield was consistently 10–15 µg of protein per milliliter of cell culture medium (data not shown), and purity was −90% (Figure 2B). When enzymatic activity was measured using the NAALADase assay described in Materials and Methods, recombinant His-PSMA exhibited a specific activity similar to that of LNCaP membranes (data not shown), with sensitivity to known specific inhibitors of PSMA/NAALADase (see below).
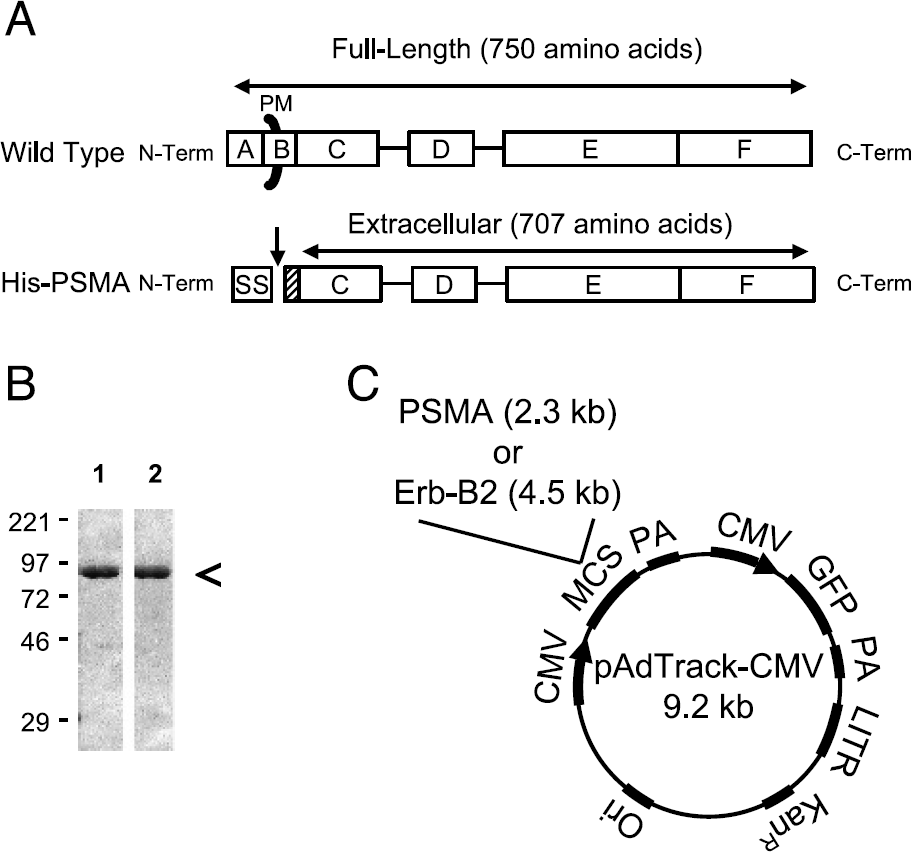
Construction of a baculovirus producing soluble PSMA and an adenovirus expressing wild-type PSMA. (A) Domain structure of wild-type PSMA (top) and recombinant His-PSMA (bottom). Domains of PSMA are as defined in the literature [40] and drawn to scale: A= intracellular, B= transmembrane, C,D= unknown function, E= catalytic, F= unknown function, PM= plasma membrane. The extracellular 707 amino acids of PSMA (amino acids 44–750) are fused to an insect cell N-terminal secretion signal (SS) and histidine tag (hashed box). The secretion signal is cleaved (downward arrow) prior to secretion into insect cell medium. (B) Gel electrophoresis of purified His-PSMA. A Coomassie-stained 10% SDS-PAGE gel with molecular weight markers (kDa) is shown. Lane 1: 2.5 µg of His-PSMA purified from insect cell medium using nickel agarose. Lane 2: 2.5 µg of His-PSMA eluted from nickel agarose using imidazole and dialyzed against TBS. (C) Production of adenoviruses co-expressing GFP and either full-length, wild-type human PSMA, or Erb-B2 using the pAdTrack-CMV vector [28]. Co-expressed proteins are under the control of independent CMV promoters. MCS = multiple cloning site, PA = polyadenylation signal, LITR = Left-sided inverted terminal repeat, KanR = kanamycin resistance, Ori = bacterial origin of replication.
PSMA/NAALADase Enzymatic Assays and Structure-Activity Relationships
To evaluate the large number of compounds used in this study, we employed the NAALADase enzyme inhibition assay described in Materials and Methods. Because PSMA's enzymatic activity requires Cl− ion and is inhibited by phosphate (Ki ≈ 100 µM [27]), all reactions were performed in Tris-HCl buffer.
As described above, there was precedence in the literature to suggest that the binding activity of β-NAAG is insensitive to removal of the acetyl group, which is demonstrated by only twofold potency decrease of β-AG (Figure 3A). Unexpectedly, conjugation of β-AG with the NIR fluorophore IRDye78 [23] to form β-AG-78 resulted in an almost 300-fold improvement in PSMA inhibitory activity to 8.1 nM (Figure 3A). For reference, also shown in Figure 3A is 2-PMPA, the most potent (0.4 nM) NAALADase inhibitor described to date [30].
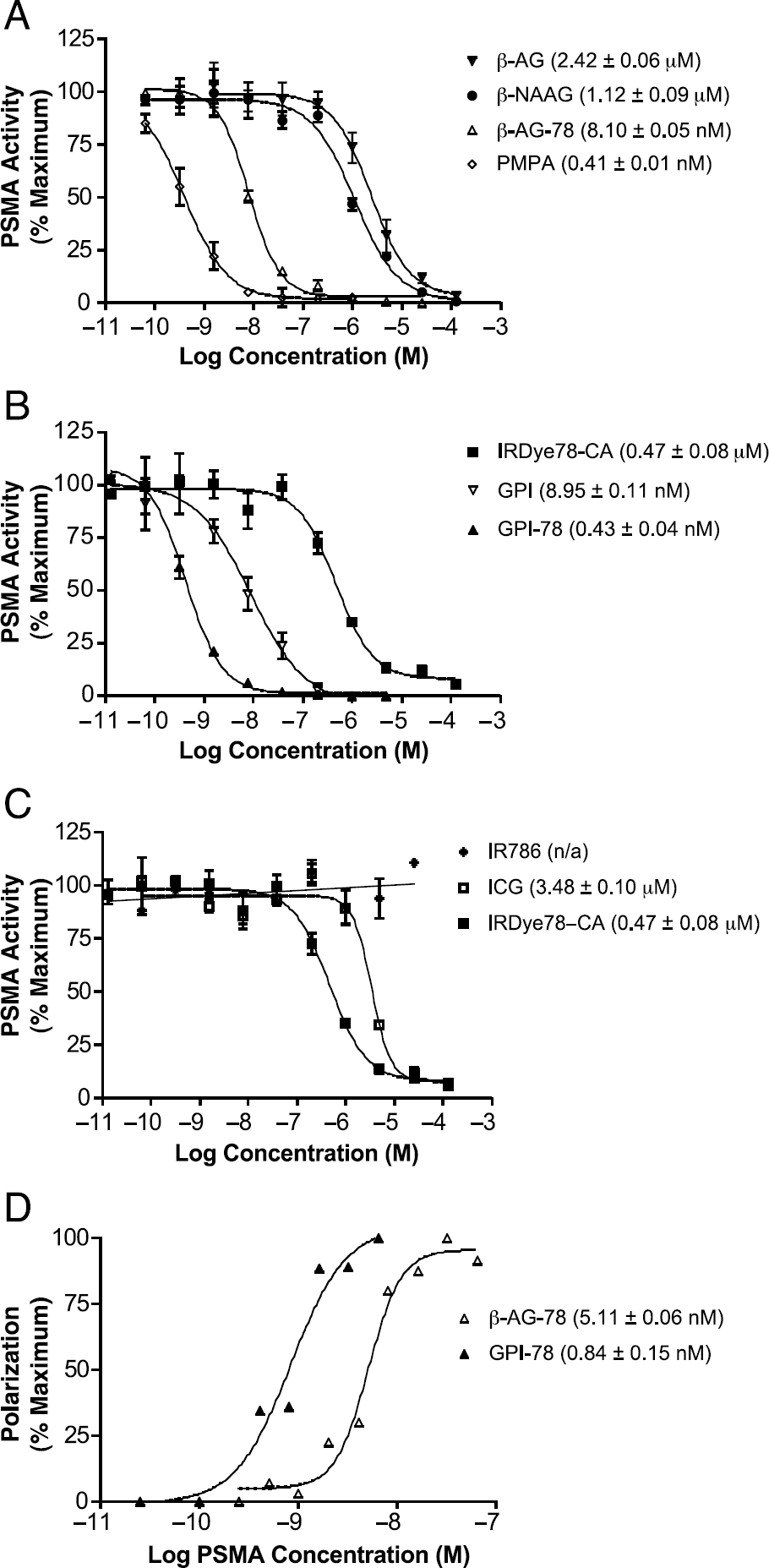
Structure-activity relationships among PSMA/NAALADase substrates, inhibitors, and ligands. PSMA/NAALADase enzymatic assays were performed as detailed in Materials and Methods. Shown are mean activities ± standard deviations from triplicate assay points. (A) Enzymatic activity in the presence of increasing concentrations of β-AG (filled reversed triangles), β-NAAG (filled circles), β-AG-78 (open triangles), and PMPA (open diamonds). (B) Enzymatic activity in the presence of increasing concentrations of IRDye78-CA (filled squares), GPI (open reversed triangles), and GPI-78 (filled triangles). (C) Enzymatic activity in the presence of increasing concentrations of IR-786 (stars), ICG (open squares), and IRDye78-CA (filled squares). (D) Ligand-binding affinity measured using fluorescence polarization. Polarization curves for 1 nM β-AG-78 (open triangles) or 1 nM GPI-78 (filled triangles) incubated with an increasing molar concentration of His-PSMA.
Although β-AG-78 exhibited a reasonable Ki, we wondered whether higher affinity single nucleophile-containing ligands could be developed and converted into NIR fluorescent contrast agents. One such compound would be the aminated derivative of compound 7 in Ref. [30]. However, already available to us was the GPI compound with a three-carbon linker [25]. As shown in Figure 3B, GPI was a potent inhibitor of NAALADase, with a Ki of 9.0 nM. Importantly, conjugation of GPI to IRDye78 resulted in an over 20-fold improvement of affinity (0.4 nM). It should be mentioned that the obtained value is actually at the limit of the NAALADase assay, and affinity may, in fact, be higher.
Intrigued by the synergy seen after conjugation, we explored whether IRDye78 itself bound to the active site of PSMA. As shown in Figure 3B, IRDye78-CA had an inhibitory activity (0.47 µM) similar to β-NAAG (1.1 µM). Because PSMA active site binding molecules tend to be highly anionic, we hypothesized that tetra-sulfonation was accounting for our observation. As shown in Figure 3C, the di-sulfonated heptamethine indocyanine ICG had a markedly reduced inhibitory activity, and the nonsulfonated molecule IR-786 [32] had no measurable inhibitory activity, suggesting that tetra-sulfonation, with the appropriate spacing, was the mechanism underlying the unexpected affinity of NIR fluorophores for PSMA. The inhibitory activities of all molecules used in this study are summarized in Table 1.
Validation of Enzyme Inhibition as a Surrogate for Ligand Binding
Although enzymatic assays are convenient to evaluate compounds, it is always possible that enzymatic inhibition does not faithfully correlate with ligand binding to the active site. For this reason, we independently evaluated the ligand-binding affinity of β-AG-78 and GPI-78 using fluorescence anisotropy. As shown in Figure 3D, the Kd of these compounds were nearly identical to the Ki of the compounds, thus validating the use of enzymatic inhibition as a surrogate for ligand binding.
Inhibitory/ Binding Constants for PSMA Active Site Molecules (in Increasing Order of Affinity)

Specificity of GPI-78 Binding to Cell-surface PSMA
Encouraged by the in vitro results, we asked whether GPI-78 was capable of specific binding of PSMA on the surface of living cells. To do so, we constructed two pAdEasy-1 [28] adenoviruses (Figure 2C), one co-expressing GFP and PSMA, one co-expressing GFP and Erb-B2, an unrelated cell-surface protein overexpressed in breast cancer. As shown in Figure 4A, GPI-78 bound to PSMA-negative PC-3 human prostate cancer cells overexpressing PSMA, but not Erb-B2. Importantly, there was complete concordance between GFP and independent immunofluorescence staining of cell-surface receptors (Figure 4A), validating the use of GFP alone as a surrogate for PSMA and Erb-B2 expression in future experiments.

NIR fluorescence imaging of endogenous and ectopic wild-type human PSMA on living cells, and the contribution of endocytosis to signal strength. (A) PSMA-negative PC-3 cells infected 48 hr previously with an adenovirus co-expressing GFP and either wild-type PSMA (top) or Erb-B2 (bottom). 2 µM GPI-78 in TBS was incubated with cells for 20 min at RT prior to washing, fixing, and mounting. Shown are NIR fluorescence (left), indirect immunofluorescence (second from left) with either an anti-PSMA (top) or anti-Erb-B2 monoclonal antibody (bottom), GFP co-expression (second from right), and phase contrast (right) images. Fluorescence images have identical exposure times and normalizations. (B) NIR fluorescence imaging of endogenous PSMA on living human prostate cancer cells was performed using PSMA-negative PC-3 cells (top) and PSMA-positive LNCaP cells (bottom). Cells were incubated with 2 µM GPI-78 in TBS for 20 min at RT prior to washing, fixing, and mounting. Shown are NIR fluorescence (left) and phase contrast (right) images. Fluorescence images have identical exposure times and normalizations. (C) Effect of endocytosis on signal enhancement. LNCaP cells were incubated with 2 µM GPI-78 (top) or IRDye78-CA (bottom) in TBS for 20 min at either 37°C (left) or 4°C (right) prior to washing, fixing, and mounting. Shown are NIR fluorescence (left) and phase contrast (right) images. Fluorescence images have identical exposure times and normalizations.
GPI-78 Binding to Endogenous PSMA and the Contribution of Endocytosis to Signal Strength
Because CMV promoter-based overexpression often leads to high protein levels with no biological relevance, we asked whether GPI-78 would detect endogenous PSMA on the cell surface. As shown in Figure 4B, GPI-78 bound to 100% of the PSMA-positive LNCaP human prostate cancer cell line, but not to the PSMA-negative cell line PC-3. Because PSMA is known to undergo both constitutive and antibody-mediated endocytosis, we quantified the contribution of endocytosis to the observed signal using NIR fluorescence microscopy (Figure 4C). For a 20-min GPI-78 incubation time, approximately 50% of the signal strength was lost when cells were incubated at 4°C versus 37°C. Consistent with this observation was a change in the pattern of cell-associated GPI-78 from cell surface and intracellular (Figure 4C, 37°C) to cell surface only (Figure 4C, 4°C). Lysis and measurement of LNCaP cells incubated with 2 µM GPI-78 at 4°C for 20 min revealed that 3.2 × 105 receptor sites per cell were occupied.
Blood Half-life and Organ Uptake of GPI-78
To be useful for intraoperative imaging, NIR fluorescent contrast agents should have rapid biodistribution and clearance. As shown in Figure 5A, both IRDye78-CA and GPI-78 were cleared rapidly from the blood; early- and late-phase half-lives for IRDye78-CA were 1.6 and 3.4 min, respectively, and for GPI-78, 1.5 and 2.3 min, respectively. These values are consistent with previously published values from our group using Pam78, a NIR fluorescent contrast agent specific for hydroxyapatite [33], although the microcapillary-based method we present in this study has a greatly improved time resolution, minimizes blood handling, and is much more accurate.
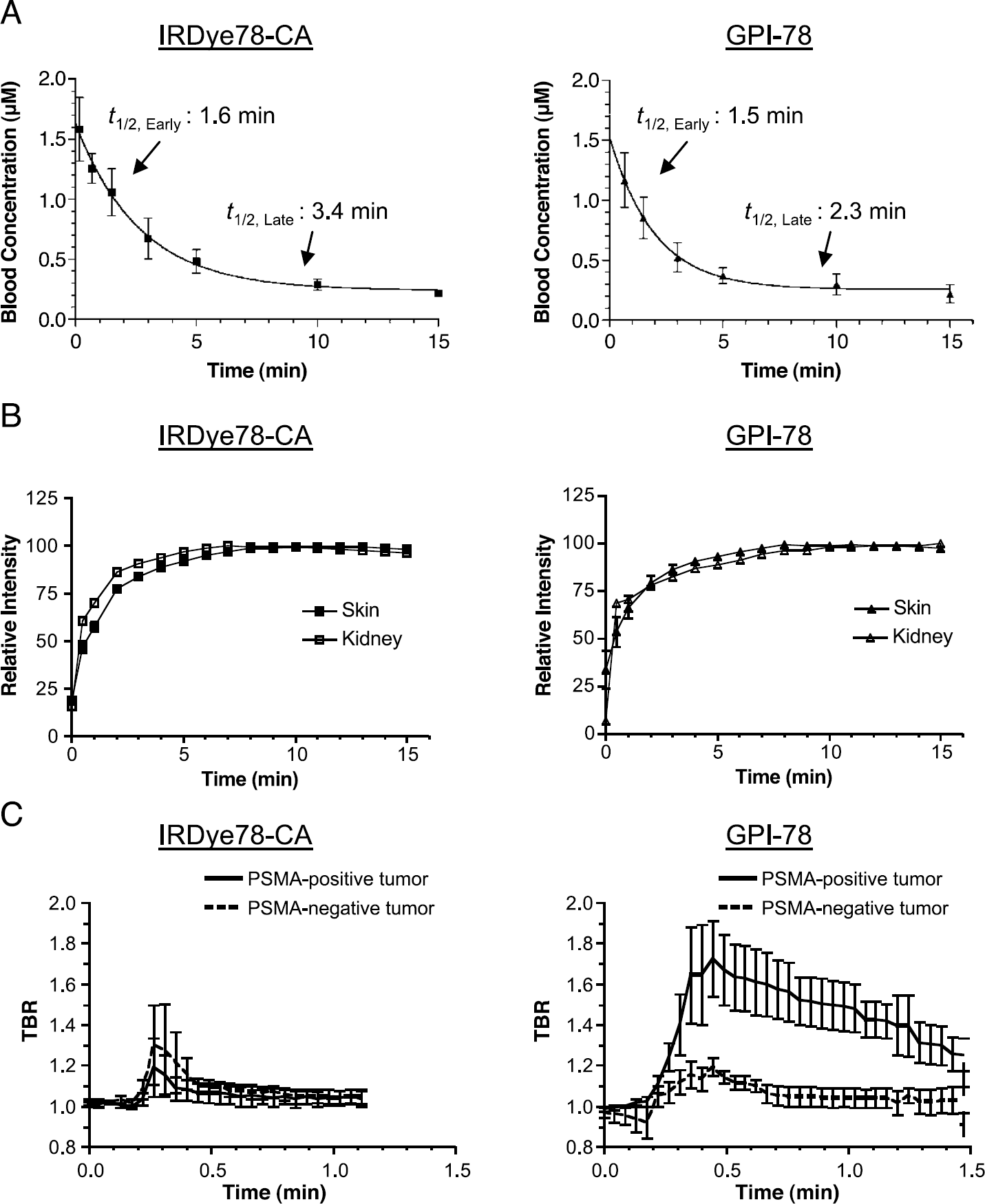
Blood clearance and organ/tumor uptake of IRDye78-CA and GPI-78. (A) Blood clearance of IRDye78-CA (left) and GPI-78 (right) as measured using the protocol described in Materials and Methods. Data points represent mean ± SEM NIR fluorescence measurements on multiple animals (n = 3). Also shown are the early-phase (t1/2, Early) and late-phase (t1/2, Late) elimination half-lives. (B) Skin (filled) and kidney (open) uptake as a function of time after injection of IRDye78-CA (squares) or GPI-78 (triangles). Shown is relative uptake. The regions of interest used for the measurements are shown in Figure 6. (C) TBR of IRDye78-CA (left) or GPI-78 (right) in PSMA-positive (LNCaP) and PSMA-negative (TsuPR1) tumors as a function of time after intravenous injection. The regions of interest used for the measurements (mean ± SEM) are shown in Figure 6. n = 5 animals for GPI-78 and n = 3 animals for IRDye78-CA.

In vivo imaging of human prostate cancer xenograft tumors in athymic nu/nu mice. Xenograft tumors derived from human cells lines were grown in 25 g male athymic nu/nu mice. On the right flank was a PSMA-positive LNCaP tumor. On the left flank was a PSMA-negative TsuPR1 tumor. Shown are representative (n= 4 animals per compound) images of color video (left), NIR fluorescence (middle), and a pseudocolored merge of the two (right) 20 sec after intravenous injection of 5 nmol (0.2 µmol/kg) of GPI-78 (top) or IRDye78-CA (bottom). NIR fluorescence images have identical exposure times (250 msec) and normalizations. Also on the merged images are the regions of interest (dotted circles) used for the quantitative measurements of the tumors, background (B), kidneys (K), and skin (S) shown in Figure 5B and C.
Coincident with blood clearance was a rapid increase in signal strength in the skin and kidney, plateauing by 5 min (Figure 5B), then decreasing to background over the course of a few hours (data not shown). PSMA-positive tumors exhibited a TBR higher than PSMA-negative tumors for GPI-78 (Kd ≈ 400 pM) but not IRDye78-CA (Kd ≈ 500 nM), with GPI-78 maintaining its TBR over the next 2 min (Figure 5C; discussed below). For injection of GPI-78, the peak TBR for PSMA-positive tumors was 1.80 ± 0.21 (mean ± SEM) and for PSMA-negative tumors was 1.20 ± 0.05. This difference was significant (p = .024) using a two-tailed unpaired Student's t test. For injection of IRDye78-CA, the peak TBR for PSMA-positive tumors was 1.20 ± 0.11, and for PSMA-negative tumors was 1.30 ± 0.21 (p = .7). At 4 hr postinjection, the total uptake of GPI-78 in LNCaP tumors was approximately 0.06% ID/g, likely representing the small mass of material endocytosed during the initial binding event.
In Vivo NIR Fluorescence Imaging with GPI-78
LNCaP and TsuPR1 xenograft tumors were grown in athymic mice. PSMA-positive LNCaP tumors exhibited NAALADase activity of 10 pmol/min/mg in the membrane fraction and 2 pmol/min/mg in the supernatant fraction. PSMA-negative TsuPR1 tumors exhibited 0 pmol/min/mg of NAALADase activity in both the membrane and supernatant fractions. As a frame of reference, LNCaP cells grown in tissue culture exhibit a specific activity of 10 pmol/min/mg. Immunoblotting was consistent with these results (data not shown). Shown in Figure 6 is in vivo imaging of PSMA-positive LNCaP and PSMA-negative TsuPR1 xenograft tumors 20 sec after intravenous injection of 5 nmol (0.2 µmol/kg) of GPI-78 or IRDye78-CA. Although kidney and skin uptake was similar among all animals studied, only PSMA-positive tumors exhibited appreciable tumor signal above background, with GPI-78 being brighter than IRDye78-CA, as would be expected from the measured affinities of the reagents (Figure 3 and in Table 1).
Discussion
This study presents essentially complete preclinical development of a small-molecule NIR fluorescent contrast agent targeted to prostate cancer, from conception, preparation of reagents, synthesis, and purification, through in vitro analysis, cellular analysis, and in vivo imaging. Each component of the study is essential for understanding the affinity, specificity, optical properties, and in vivo properties of the agent.
Real-time intraoperative imaging during cancer surgery has the potential to greatly improve patient care. First, the type of color-NIR merge image shown in Figure 6 and in many previous publications from our group (e.g., Ref. [34]) provides the surgeon with tumor location in relation to surgical anatomy. This is of critical importance as the NIR fluorescence image alone is not very useful intraoperatively. Alternatively, the NIR fluorescence channel can be used to highlight blood vessels [22] or other critical structures during cancer resection to prevent patient morbidity. For prostate cancer surgery, the combination of intraoperative NIR fluorescence imaging system and a targeted NIR fluorescent contrast agent could be used to highlight the boundaries of the gland, to find small collections of tumor cells in-transit to pelvic lymph nodes, and possibly to assess pelvic lymph nodes in real time. One might also imagine using such an agent preoperatively for guiding transrectal biopsy.
Two key features of a targeted NIR fluorescent contrast agent for image-guided surgery are: (1) the ability to repeat administration after initial cancer resection in order to provide high sensitivity inspection of the surgical field and to eliminate photobleaching as a source of false-negative imaging and (2) to provide high signal strength. To achieve the former, our laboratory focuses on the development of low-molecular-weight ligands. Because of their small hydrodynamic diameter, such ligands exhibit rapid biodistribution, rapid clearance, and potentially improved tumor uptake. The time required to achieve a high TBR is especially important in an operating room environment where charges are often accrued on a per-minute basis. To achieve the latter, our laboratory has carefully analyzed photon transmission in Rayleigh-dominant and Mie-dominant tissues of varying hemoglobin-to-water ratios [35], and have concluded that organic fluorophores in the 800-900 nm range are advantageous. To achieve such wavelengths while maintaining high QYs, we typically utilize tetra-sulfonated heptamethine indocyanines, such as IRDye78. We are also extremely careful when handling these fluorophores to avoid degradation [23].
Tumor-specific ligands that target the active site of an enzyme are attractive because they can often be reengineered to include a strong nucleophile or other chemical group for covalent conjugation to contrast agents. Indeed, several classes of small-molecule PSMA inhibitors have been described [25,30,36–38], one of which was the basis for the present study. However, our data highlight the major pitfall with this approach. Endogenous ligands and substances will often compete for the very site being targeted. This is especially worrisome for hormone derivatives, where receptor occupation can change during normal physiological cycles.
Although we describe a molecule with subnanomolar affinity, a molecular weight of only ≈1400 Da, 800 nm fluorescence, and PSMA-specific binding, we are clearly not satisfied with the in vivo results. As mentioned above, all in vitro experiments, including cellular studies, were conducted in Tris-HCl buffer because phosphate competes with the active site of PSMA (Ki ≈ 100 µM). In Tris buffer, cellular binding of GPI-78 was rapid, specific, and likely complete, with approximately 3.2 × 105 occupied receptor sites per cell. In vivo tumor uptake was rapid, specific, but transient, despite tumor immunoblots that confirmed PSMA expression during xenograft growth (data not shown). The most likely explanation is that the relatively low blood concentration of GPI-78, approximately 3 µM after initial intravenous injection, and its high off-rate, cannot effectively compete with endogenous phosphate (≈1.2 mM), especially because GPI-78 blood clearance is extremely rapid (Figure 5A). This also helps explain results from another group, in which experiments conducted in phosphate-containing cell culture medium showed an unexpectedly low number of binding events after aptamer-based targeting to PSMA's active site [19]. Our data also call into question whether PSMA expression on the neovasculature of human solid tumors [8,9] results in an active extracellular enzyme, an enzyme that is only active in the cytosol as the truncated PSM' form [7], or a protein that is merely a marker for some other genetic change.
Tumor-targeted in vivo imaging with contrast agents often fails because of the many physiological barriers that prevent an adequate TBR from being attained. One such barrier, discussed above, is the presence of endogenous substances that compete with the contrast agent. Another is the tight junctions of many tissues. The latter is of paramount importance for PSMA imaging. The highest levels of PSMA are in the brain, lumen of the jejunum, and prostate. From an imaging perspective, the former two sites are “sanctuary sites” as either tight junctions or a mucosal basement membrane prevents distribution of contrast agent. In the prostate, PSMA has been localized to the secretory acinar cells of the gland. One must wonder, therefore, what fraction of normal prostate cells is accessible to intravenously injected contrast agents. If, as we suspect, only a small fraction of PSMA-positive normal cells are accessible, then contrast agents targeting PSMA might provide a higher-than-expected specificity for malignant over normal glands. An unresolved question is the accessibility of PSMA in the kidney. In our studies, there was no difference in kidney signal between IRDye78-CA, β-AG-78, and GPI-78 in either wild-type mice, or transgenic mice homozygously null for PSMA (data not shown). This result brings into question whether renal PSMA is accessible, even to a filtered small molecule, and whether kidney uptake is truly a surrogate for PSMA binding [14].
It should also be emphasized that receptor imaging with PSMA is likely a best-case scenario. Even with 3.2 × 105 occupied receptor sites per cell in the absence of endocytosis (consistent with previously published values [16,17]), the absolute contrast agent concentration over the volume of a typical cell (1-2 pL) is only 250–500 nM. For most other receptors, the achievable contrast agent concentration will be one to three orders of magnitude lower. Although the former concentrations are within the realm of in vivo fluorescence-based imaging, it remains to be seen just how low cell-surface receptor density can be before fluorophore emission is overwhelmed by tissue autofluorescence, and so forth.
We acknowledge two major problems that could limit the usefulness of GPI-78. First, our data suggest that the hydrodynamic diameters of some NIR fluorescent contrast agents might actually be “too small” for effective imaging. That is, renal clearance is so rapid that tumor contact time is minimal. To solve this problem, the GPI ligand could be conjugated to a larger NIR fluorescent carrier molecule. However, the second problem is competition with endogenous anions such as phosphate. A potential solution to this problem is to combine an increase in hydrodynamic diameter with multivalency. As has been shown so elegantly by the Whitesides group (reviewed in Ref. [39]), multivalency is a common strategy in nature to decrease ligand off-rate and improve ligand affinity. Whether a multivalent GPI-78 derivative will improve in vivo performance is the subject of continuing experimentation.
Footnotes
Acknowledgments
We thank Daniel R. Draney, PhD (LI-COR) for IRDye78-CA, James K. Coward, PhD (University of Michigan) for an authentic sample of GPI, Joseph T. Coyle, MD (McLean Hospital) for plasmid PSMA2, Cary H.C. Lai, PhD (Scripps Research Institute) for vector pAcGP67BHis, Kermit Carraway, PhD (UC Davis) for the Erb-B2 cDNA, John T. Isaacs, PhD (Johns Hopkins) for the TsuPR1 human bladder cancer cell line, Barbara L. Clough for editorial assistance, and Grisel Rivera for administrative assistance. This work was supported by grant R21/R33-CA-88245 (JVF) from the Cancer Imaging Program, National Cancer Institute, National Institutes of Health.