
Abstract
Ischemic stroke is emerging as a major health problem for elderly women. Women have lower stroke incidence than men until an advanced age, when the epidemiology of ischemic stroke shifts and incidence rises dramatically in women. Experimental models of rodent stroke have replicated this clinical epidemiology, with exacerbated injury in older compared with young female rodents. Many of the detrimental effects of aging on ischemic stroke outcome in females can be replicated by ovariectomy, suggesting that hormones such as estrogen play a neuroprotective role. However, emerging data suggest that the molecular mechanisms leading to ischemic cell death differ in the two sexes, and these effects may be independent of circulating hormone levels. This article highlights recent clinical and experimental literature on sex differences in stroke outcomes and mechanisms.
Keywords
Stroke is the third leading cause of death in the USA and the leading cause of disability [1]. Women enjoy a lower stroke incidence relative to men until an advanced age [2], when this incidence reverses leading to peak stroke risk in women aged over 80 years. The majority of strokes occur in the aging population. By the year 2050, approximately one in five Americans will be elderly, with a disproportionate number of elderly women represented [301]. Therefore, it is the aging female population that will bear the major burden of stroke-related disability and institutionalization [3].
For many years, the shift in ischemic risk in women has been attributed to the loss of hormones, especially estrogen, at the time of menopause. Earlier menopause (natural or surgical) leads to accelerated atherosclerosis, which is a risk factor for stroke [4]. Epidemiological evidence from the Framingham study confirms this, as an increased risk of ischemic stroke was seen in women who had an earlier age (42 years) of natural menopause [5]. This risk has been modeled in preclinical stroke studies in female rodents and primates that have found exacerbated tissue damage after ‘surgical menopause’ (ovariectomy). Despite the strong evidence that gonadal hormones protect in induced stroke models [6], clinical trials have failed to translate the neuroprotective effects of estradiol into a viable therapy for stroke. Incorrect dosing and the timing of estrogen replacement have probably played a role in this translational failure, but other, less understood, hormone-independent factors may also play a role. The loss of ovarian function with the cessation of estradiol production occurs much earlier in life (early 50s) than the increase in ischemic sensitivity in women (age over 80 years), suggesting that there are other contributing factors. It is increasingly recognized that the cell death pathway initiated by experimental ischemic insults can differ based on the sex of the cell or animal examined, regardless of the hormone status. Genetic sex (XX versus XY) and epigenetic modifications have their own critical role to play. This article focuses on the contributions of basic science towards our understanding of the pathophysiology of stroke in males versus females.
Sex differences in clinical stroke Epidemiology
Clinically, it has been well recognized that sex differences exist in ischemic stroke. These differences are diverse, ranging from differences in epidemiology, stroke prevention, acute stroke treatment and outcomes [7,8]. Men have a higher incidence of stroke compared with women until an advanced age [1], after which rates rise dramatically in women. The Framingham study demonstrated that the reversal in stroke sensitivity in women occurs after the age of 85 years [9]. Similarly, a Swedish and Oxford Study demonstrated lower stroke incidence for women aged 55–74 years compared with age-matched men, which reversed for women aged 85 years and older [10]. However, this epidemiology may be changing with the increase in obesity and metabolic syndrome in middle-aged women. The American Heart Association (AHA) 2011 update reported that the risk for incident stroke at 65 years of age has decreased significantly from 19.5 to 14.5% in men, but more modestly from 18.0 to 16.1% in women [11]. In the National Health and Nutrition Examination Survey (NHANES) conducted by the CDC for the years 1999–2004, it was found that women 45–54 years of age had twice the odds of having experienced a stroke compared with men in the same age group [12]. This increase in incidence in middle-aged women may also be attributed to pregnancy-related physiologic and hemodynamic changes as maternal age has also increased. Ecclampsia accounts for 24–47% of ischemic strokes during pregnancy or puerperium and contributes to the incidence of stroke in younger middle-aged women [13,14].
Despite the higher overall incidence of stroke in men throughout the lifespan, women account for 60.6% of stroke deaths [1]. Although it may be argued that the increased mortality may be due to the higher age of first stroke in women (74.5 vs 69.2 years), functional recovery after stroke and stroke severity is also worse in women, even in age-normalized data. In a 2005 study, at 3 months poststroke only 13% of women performed eight of the nine instrumental activities of daily living versus 28% of men; and at 6 months poststroke 18% of women could perform these activities versus 34% of men [3]. The Barthel Index Score (with higher scores reflecting improved function) was higher for men (18.5 ± 3.6) than women (16.7 ± 5.5), after stroke [15]. This gender gap not only persists, but appears to widen over time, as sex differences were even larger at 12 months compared with 3 months poststroke [16].

Modified Barthel Index, for males and females at four time points – 1: before admission; 2: at the time of admission; 3: at 3 months poststroke; and 4: at 12 months poststroke.
Management
Disparities exist in the acute management of stroke, and much of this data has been recently reviewed [7,8,21]. In Germany, women were less likely to be admitted to the hospital within the first 3 h of stroke onset and had a 13% lower chance of receiving thrombolytic therapy [22]. However, other groups have found that there are no sex-related differences in the rates of administration of tissue plasminogen activator [23–25], possibly owing to increasing awareness or geographic variations. A recent report from the Mechanical Embolus Removal in Cerebral Ischemia (MERCI) trial reported that although women were older and more likely to have hypertension, no sex differences existed in revascularization rates or clinical outcomes after mechanical embolectomy [26]. Lack of an effect of sex was also seen in the basilar artery acute occlusion study [27]. The Brain Attack Surveillance in Corpus Christi (BASIC) study found that women were less likely to undergo carotid artery evaluation (71% males vs 62% females) or echocardiography (57% men vs 48% women) [28]. The older age of women with stroke may be one possible reason for these sex differences, or evidence of a clear stroke etiology that does not necessitate further work up (i.e., known atrial fibrillation with subtherapeutic anticoagulation). In addition, although women with ischemic stroke or transient ischemic attack are less likely than men to undergo carotid screening and revascularization, this difference is largely explained by potential contraindications to surgery and by lower severity of carotid disease in women [29].
Treatment rates with antithrombotic medications for secondary prevention of stroke are known to decrease with age and are lowest for patients greater than 85 years of age, which would disproportionately affect women [30]. The Registry of the Canadian Stroke Network (RCSN) reported that the sex differences seen in care in an acute stroke unit were no longer significant after adjustment for age and other factors [18]. A study by Gall et al., in 2010, using the North East Melbourne Stroke Incidence Study (NEMESIS) data reported that the sex differences seen in their study could be explained by the severity of stroke in women, the higher number of comorbidities and older age at presentation [31]. Patients living in dependent care before stroke were less often admitted to the hospital than those who lived independently. Therefore, although barriers exist to the access of healthcare to women, the magnitude of these differences may be small after adjusting for age and other factors such as social frailty [31].
Other contributing factors to sex differences in acute management may include differences in clinical stroke presentation [32], leading to a lack of recognition of stroke symptoms by the patient or healthcare provider. However, most studies demonstrate that most women present with ‘typical’ stroke symptoms similar to men [33–35], arrive to medical attention with the same speed as men, and are recognized without delay [16]. Women have a surprisingly low perception of their risk for stroke, even when they have known risk factors for stroke such as hypertension and atrial fibrillation [36]. The existence of sex differences in stroke is most likely due to a multitude of these interacting factors, some of which are often not controlled in large epidemiological databases. Risk factors that play a unique role in women, including central adiposity, endogenous sex hormones and psychological factors, such as depression, are also emerging [37] and merit further study. Clinical research studies that evaluate the sexes separately are desperately needed. Until the underlying reasons for these sex differences are discovered, the burden of stroke-related disability in older women will continue to grow.
Sex differences in clinical stroke.

Coagulation & fibrinolysis
In a pooled analysis of thrombolytic trials [38,39], women had a greater margin of benefit compared with men when treated for stroke with thrombolytic drugs compared with placebo. Women who received placebo had worse outcomes than men in each of these trials, increasing the margin of benefit of treatment in women. The same effect was seen in an analysis of a registry of routine stroke care [40]. Using noninvasive vascular imaging to assess vessel patency within 72 h of intravenous tissue plasminogen activator treatment, it was found that any recanalization (TIMI score: 1–3) was more likely in women after thrombolysis [41]. Sex differences in fibrinolysis and coagulation have only recently been evaluated. Aging and female sex have significant procoagulant effects [42]. For instance, the concentration of plasminogen activator inhibitor (PAI)-1 (a procoagulant) increases postmenopause [43], which could increase the risk of thrombosis in women. Higher levels of PAI-1 were seen in patients with acute stroke, and higher plasma PAI-1 levels were associated with thrombolysis failure [44]. Venous thrombosis rates may also differ by sex. The occurrence of deep venous thrombosis was higher in women after intracerebral hemorrhage in Japan [45], but the reverse has been seen in US populations [46]. Future studies are urgently needed in this area in both patients and animals as our use of reperfusion therapies continues to grow.
Sexual dimorphism in experimental stroke
Background: experimental models of stroke
Stroke can be modeled in the laboratory using in vivo or in vitro techniques. In vitro, ischemic stroke is most commonly modeled by depriving brain slices or neuronal cultures of oxygen and glucose (oxygen glucose deprivation; OGD model) although oxidative stress and NMDA excitoxicity have also been used as in vitro models [47,48]. Sex differences have been seen in organotypic brain slices [49], as well as in neuronal [50], astrocyte [51] and, recently, endothelial cell cultures [52]. Although in vitro stroke modeling is technically quite straightforward, cost effective, can elucidate the specific cell type contributions to the ischemic response (i.e., astrocyte vs neurons) and allows genetic manipulations (knockdown of specific proteins of interest) that are often impossible to perform in vivo, these methods do have several limitations. Cell culture models do not maintain the normal in situ interactions between the vascular and cellular (neuronal, microglia and astrocytes) components of the brain that are so important to the ultimate response in a patient with stroke. Many scientists have tried to modify the in vitro model of OGD to make it more translational and utilize neurons cocultured with microglia or astrocytes prior to injury induction [53–55]. Recently, Richard et al. devised a novel method by which focal ‘ischemia’ can be induced in the OGD model by applying OGD medium focally to a small portion of a brain slice while bathing the remainder of the slice with normal oxygenated media [56]. This led to electrophysiological changes in the infarct area, with a reduction in synaptic transmission within 50 min of OGD medium application. Although these models are extremely useful in the elucidation of cellular events that are difficult to assess in the living animal, often immature brain slices and neurons are utilized, making the findings less translationally relevant to the population at risk for stroke – older adults. It is unlikely that the complexity of the brain and its vasculature, especially when considering effects of aging and sex, can be evaluated in an in vitro system [57].
There are numerous in vivo experimental models of stroke in common use in the laboratory [58]. These include both focal (one vessel) and global (hypoxia of the entire brain, which models cardiac arrest in humans). The occlusion of vessels can be achieved by photothrombosis (photothrombotic model), inserting an intraluminal suture (intraluminal middle cerebral artery occlusion [MCAO] model), injection of a vasoconstrictor into the vessel or directly into the brain (endothelin-1 model) or by introducing emboli in the blood vessels (embolic model) [58,59]. The injection of autologous thrombi/emboli into extracranial arteries that subsequently reach the more distal intracranial arteries has been increasingly used as a more translational model of stroke in humans. However, the model can be difficult to perform in small rodents and the timing of reperfusion, if it occurs, is difficult to control [60]. As 51% of strokes are caused by occlusion of the middle cerebral artery (MCA) [61], and reperfusion therapies are offered to an increasing number of patients (now 28% at our stoke center at Hartford Hospital) transient focal occlusion of the MCA is an important and relevant animal model. This model allows for reversible occlusion of the MCA, the duration of which can be varied (by removing the intraluminal suture after a certain set time point) and the study of reperfusion injury. However, permanent models of focal cerebral ischemia (where suture is not removed) have also been used with success, and studies should be performed in both to confirm results prior to embarking on expensive clinical trials.

Vascular anatomy at the base of the mouse brain.
Stroke varies in its etiology and severity in individual patients. Outcomes vary depending on age, comorbid disease states, risk factor profiles and baseline functional status, as well as sex. Most animal models do not account for these complex interacting factors. Nevertheless, attempts have been made to model some of these risk factors at the bench, including the use of apoE-/− mice with accelerated atherosclerosis [62], stroke-prone spontaneously hypertensive rats [63], and models of diabetes. Interestingly, the latter models exhibit the same sex differences seen in wild-type strains without these risk factors (see below and review [64]). Only recently have sex differences in aged animals been addressed, and the results are quite variable (see later).
Sex differences in experimental models of stroke
Epidemiological studies demonstrate an intrinsic male ‘ischemic sensitivity’ or an intrinsic female ‘ischemic protection’ phenotype, at least until the age of 65 years. Similar epidemiological findings have been seen in pediatric stroke, with boys having higher incidence and poorer outcomes after stroke [2,65,66], suggesting that circulating hormone levels cannot completely account for sex differences. Sex differences have also been seen in both in vitro and in vivo models of cerebral ischemia and neuronal injury. These findings have been recently reviewed [67]. Neurons derived from females (XX) tolerate toxic dopamine exposure and have a survival advantage compared with male-derived neurons (XY) [68,69]. Female neurons were resistant to nitrosative stress compared with males, suggesting a differential molecular sensitivity to cell death triggers in male and female cells (XX vs XY), at least in vitro [50]. Similar findings of intrinsic female protection have been seen in hippocampal slice models and cultured astrocytes after OGD [49,70]. In vivo studies in young animals mirror these findings (see review [71]).
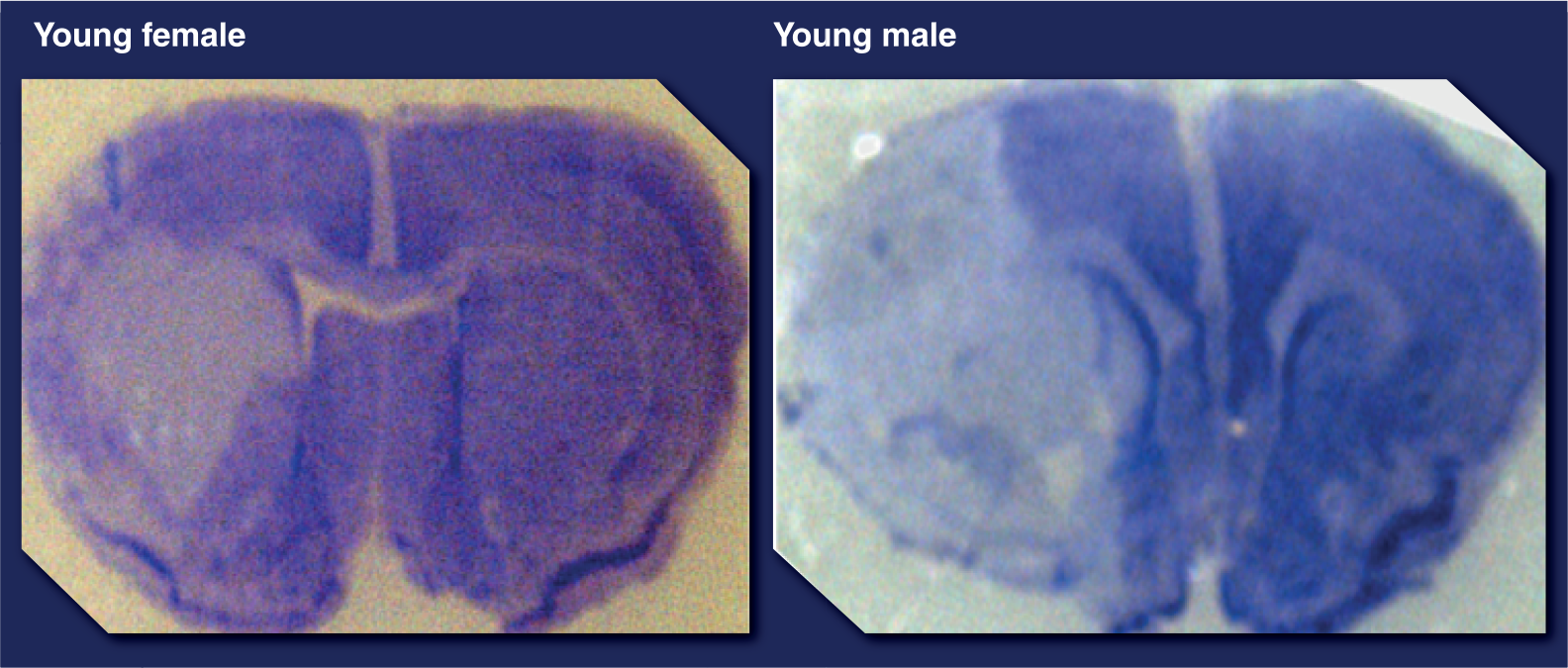
Cresyl violet staining of coronal sections of brains 24 h after transient middle cerebral artery occlusion in young (8 weeks) male and female mice.
This intrinsic protection in female brain has been largely attributed to the neuroprotective effects of estradiol as acute surgical ovariectomy or low estrous states ameliorate this effect [72,73]. One key study in spontaneously hypertensive rats followed animals into old age (40 weeks) and demonstrated significantly less vascular damage and lower rates of cerebral hemorrhage in females until an advanced age. Females also had a prolonged life expectancy [74]. Similar findings have been seen in other strains developed to model common stroke risk factors [72,75,76]; females of these strains are less sensitive to a controlled ischemic insult than males. Certainly, circulating hormones play a major role in experimental models utilizing young animals (see section on ‘The role of hormones’ for a detailed insight into the roles of estrogen and testosterone); however the response in the aged brain appears to be quite different.
Disparities in the aging animal models of stroke
The few preclinical focal stroke studies that have been performed in aged animals have primarily evaluated male animals. These have shown increased, decreased or equivalent stroke damage with aging [77–79], reflecting the variability in animal models and how little is known regarding the molecular changes that occur in the aged brain. Even less is known regarding the response to stroke in the aged female brain. One study found increased edema, enhanced blood–brain barrier breakdown and larger strokes in aged (18 months) compared with young female rats [77]. Studies in middle-aged female animals, designed to examine infarct size after gonadal senescence, invariably demonstrate more severe damage in middle-aged animals compared with younger animals with intact gonadal function after experimental stroke [80–82], although there is a considerable debate as to whether 17β-estradiol replacement can reverse this phenotype. Only one study, performed in our laboratory, examined infarct size after an induced focal stroke in young and aged mice of both sexes simultaneously [78]. Reminiscent of findings from human epidemiology [78,83] we found that young ovary intact females had smaller strokes than aged females or age-matched males, but aged females had the poorest stroke outcomes, with evidence of enhanced blood–brain barrier breakdown

Evans blue extravasation showing blood–brain barrier breakdown in brains of young and aging mice of both sexes after 24 h of stroke.
Stroke-induced cell death mechanisms are sexually dimorphic
The lack of adequate oxygen and nutrient supply leads to energy depletion, triggering acute necrosis and the formation of a core infarct [90]. In addition, a delayed programmed cell death (apoptosis) occurs during briefer periods of ischemia, or in ‘penumbral’ areas [91], which maintains a low but not absent, level of cerebral blood flow. This apoptotic cell death is a promising target for protection, as is the delayed inflammatory response to injury, as time windows for treatments are probably longer than those of acute reperfusion therapies. There are several pathways to apoptotic cell death and these most likely interact at many levels. Intrinsic apoptotic cell death is triggered by mitochondrial dysfunction, and can lead to the mitochondrial release of cytochrome c with subsequent activation of the caspase cascade (‘caspase-dependent’ cell death). In addition, overactivation of the DNA repair enzyme poly-ADP-ribose polymerase (PARP)-1 leads to NAD depletion, formation of toxic PAR polymers and the subsequent translocation of apoptosis-inducing factor (AIF) from the mitochondria to the nucleus causing ‘caspase-independent cell death’. Although these pathways are not completely distinct, especially with severe ischemic injuries, it appears that caspase-dependent cell death predominates in females, and caspase-independent cell death predominates in males
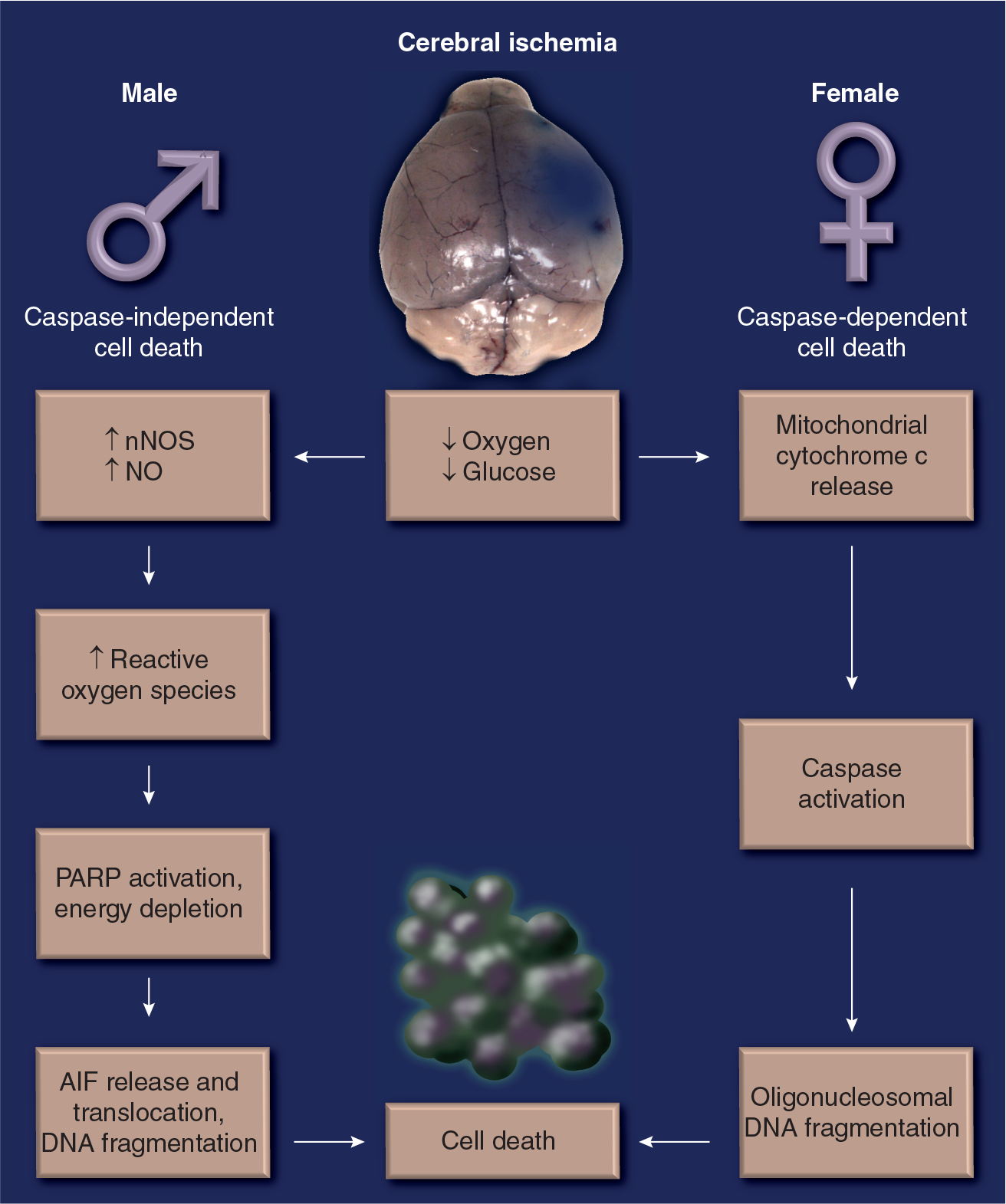
Sexually dimorphic pathways of cell death after cerebral ischemia.
Caspase-independent cell death predominates in males
Overstimulation of NMDA receptors plays a prominent role in ischemia [92]. Calcium-dependent cytoplasmic proteins are activated via the enhanced calcium influx through the NMDA receptor [93–95]. This leads to enhanced activation of neuronal nitric oxide synthase (nNOS) accelerating the production of nitric oxide. Nitric oxide combines with superoxide to form peroxynitrite, initiating DNA damage [96,97] and the activation of the DNA repair enzyme PARP-1 [98–100]. PARP-1, in an effort to repair the genome, poly(ADP) ribosylates nuclear proteins using NAD as the substrate depleting the already precarious energy reserves of the ischemic brain [101]. AIF translocates from mitochondria to the nucleus, leading to DNA fragmentation and apoptotic cell death [101–103]. Male animals with targeted deletions of nNOS, PARP or AIF have smaller infarcts after MCAO, but these manipulations either have no effect or exacerbate damage in females [104]. Similar effects have been seen with pharmacological inhibitors, such as 7-NI, a pharmacological inhibitor of nNOS, or PJ-34, a PARP inhibitor (see

Evidence for differences in cell death pathways in males and females.
Caspases are mediators of cell death in females
If females are not sensitive to PARP-mediated cell death, how do female neurons die? Evidence from in vivo models of neonates, adults and aged mice exposed to ischemic insults suggests that cell death is triggered by activation of the caspase cascade [67,109,110]. The hallmark of caspase-dependent cell death is the mitochondrial release of cytochrome c. Cytochrome c translocates to the cytosol and interacts with apoptotic protease activating factor (Apaf)-1, a CED-4 homolog and deoxyadenosine triphosphate, leading to the formation of the ‘apoptosome’ [111–115]. This activates the initiator caspase 9, with subsequent activation of executioner caspases, including caspase 3, which then cleaves the inhibitor of caspase-activated deoxyribonuclease, activating caspase-activated deoxyribonuclease and damaging DNA [116,117]. Stroke-induced cytochrome c release is higher in females versus males [109], as is caspase 3 cleavage. Similar results have been found in cortical neurons in vitro, even when hormones are removed from the culture media [50]. This implicates caspase activation as a key trigger of ischemia-induced cell death in females.
At this point, it is unknown if sex differences exist in other more recently recognized cell death pathways, such as autophagy [118] or cell death receptor signaling (Fas ligand), but these studies are currently underway in several laboratories. Possible sex differences in the response to selective inhibition of components of these pathways should be considered when developing and interpreting clinical trials.
Etiologies of sex differences in stroke: impact of genetics, epigenetics & hormones
Although the existence of sex differences in stroke is becoming increasingly accepted, the mechanisms underlying these differences and their clinical importance remain a subject of debate. Recent studies from both the clinic and the laboratories have suggested several important contributing factors to sex differences, three of which will be discussed here: gonadal hormones, genetics and epigenetic factors
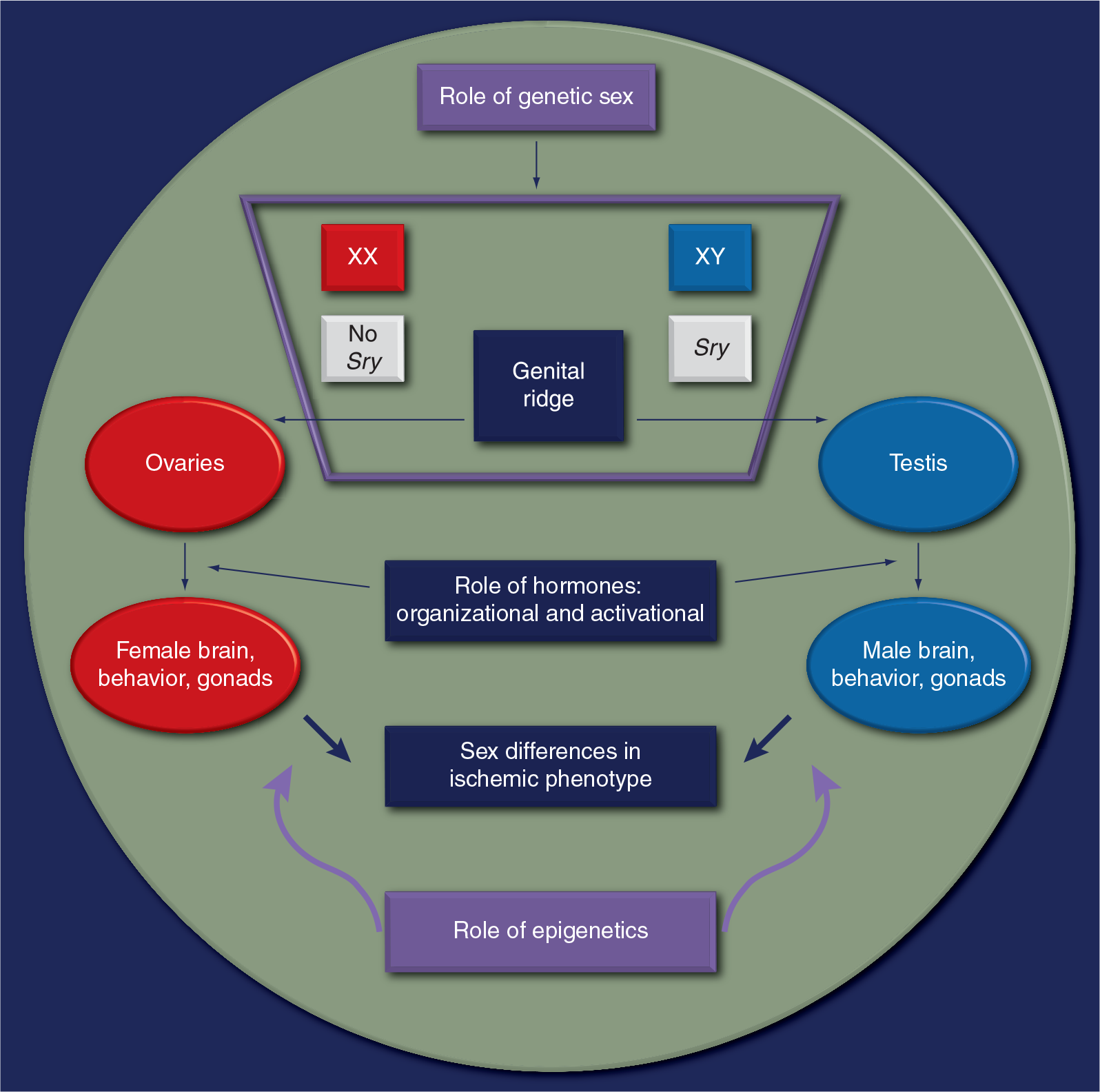
Etiology of sex differences in stroke: an interplay of hormones, genetic sex and epigenetics.
The role of hormones
The major sex hormones, estrogen and testosterone, can have both organizational and activational effects [119]. Organizational effects of hormones are permanent and independent of acute circulating hormone levels and, therefore, cannot be reversed by gonadectomy. For example, the formation of the sex-specific genitalia occurs early in life and remains stable despite changing hormonal concentrations during the lifespan. Organizational effects occur prenatally or in early postnatal life when the levels of testosterone peak in males [120]. Interestingly, this testosterone is aromatized to 17β-estradiol in the male brain, and it is this 17β-estradiol that ‘organizes’ the brain to become male as reflected by the development of sexually dimorphic nuclei and male specific behavior [121]. Males exposed prenatally to aromatase inhibitors have been shown to exhibit female specific sexual behavioral patterns (lordosis) [122]. The organization of the female brain occurs owing to the lack of estrogens as local estrogen produced by the ovaries is bound to α-fetoprotein and does not reach the brain [123].
Activational effects of hormones are dependent on the continued production of gonadal hormones and are therefore reversible by gonadectomy [119]. The interaction between organizational and later activational effects may also occur. The brain is programmed by organizational effects of hormones very early in life, but the activational effects of hormones can subsequently ‘activate’ these traits. However, activational effects are often, but not always, constrained by earlier organizational effects. For example, the male brain becomes ‘organized’ by estrogen (17β-estradiol) but the spine density in the CA1 regions of the hippocampus does not respond to the administration of 17β-estradiol in adulthood [124]. Instead, the hippocampal spine synapse growth in castrated adult male rats can be induced by androgens [125]. Conversely, the female brain, which gets organized in the absence of 17β-estradiol, has increased apical spine density in the CA1 region of hippocampus with the administration of estrogen (17β-estradiol) [124].
Estrogen-mediated neuroprotection
Estrogen (specifically 17β-estradiol) has been recognized as a neuroprotectant in most animal models of stroke [126], and most experimental studies suggest that this is an activational effect, as estradiol-mediated neuroprotection is ameliorated by ovariectomy [73,127,128]. Estradiol administered at reperfusion decreases infarct volumes in ovariectomized females and reproductively senescent females [78] as well as in males [129], suggesting that its protective effects are independent of sex and organizational factors, at least when administered acutely after injury. Estradiol's effects can be genomic (involving binding of the estrogen receptor and classic transcription after steroid/ligand binding) or nongenomic (rapid effects at the cell membrane) and its neuroprotective actions are multifaceted and still poorly understood [51,130]. Nanomolar concentrations of estradiol have been shown to be anti-inflammatory and can suppress the activation of microglia, iNOS expression and several other proinflammatory cytokines [131–135]. Estrogen can also increase neurogenesis by enhancing neuronal proliferation [136] and migration in both the subventricular zone and dentate gyrus in both sexes [6]. Postmenopausal women lose these multifactorial effects of estrogen and this may be one possible reason why they have poor functional recovery poststroke.
Estrogen is produced from testosterone via the action of aromatase, a key enzyme that mediates estrogen-mediated neuroprotection in animal models of stroke and in astrocyte culture models [70]. Local production of estrogen can occur in the brain after injury [72,137] via an upregulation of aromatase [138,139]. Female mice with genetic deletion of P450 aromatase have significantly larger infarcts than wild-type ovariectomized females, suggesting a role of local brain estradiol in neuroprotection [140]. Loss of aromatase also reduced stroke-induced neurogenesis and prolongs the duration and severity of poststroke functional deficits, which occurs in both male and female mice, suggesting a hormonal effect, rather than a sex effect, of estrogen on stroke-induced neurogenesis [89]. This can also be observed in non-injury models. Females in proestrous (with the highest estrogen levels of the estrous cycle) have higher levels of basal neurogenesis than males [141]. This is reversed by ovariectomy and partly by administration of the nonselective ER antagonist ICI 182,780 [142]. Acute administration of 17β-estradiol initially enhances, but subsequently decreases, ‘cell proliferation’ in the dentate gyrus of adult female rodents. In males, estradiol has a moderate effect on cell proliferation but instead leads to an increase in cell survival of the newly proliferated cells when administered during a specific stage in the cell maturation cycle [141]. Thus, the mechanisms of estradiol-mediated neurogenesis in adult rodents differ in males (cell survival) and females (cell proliferation). Moreover, it appears that 17β-estradiol has a primarily sex-independent effect on poststroke neurogenesis, enhancing new neuron development in mice of both sexes after MCAO.
Despite robust experimental evidence that estradiol is neuroprotective, chronic Premarin® (a conjugated equine estrogen) supplementation in postmenopausal women led to a higher incidence of stroke as well as higher rates of fatal stroke in the Women's Health Initiative (WHI) clinical trial [143,144]. In addition, the WHI Memory Study (WHIMS), an ancillary study of the WHI, reported that a combination of Premarin and medroxyprogesterone acetate given to postmenopausal women aged 65 years led to an increased risk of probable dementia [145]. Acute estradiol administration in high-risk women (with recent transient ischemic attack or nondisabling stroke suggestive of unstable plaque), also led to a significant increase in fatal stroke and enhanced stroke-induced disability [146] after treatment. There have been a multitude of explanations proposed for these findings, including independent effects of aging (laboratory studies have used young animals, whereas the average age of women in the WHI was 63 years) and confusion regarding reductions in stroke incidence (with reduction in the number of strokes with chronic use) versus acute neuroprotection (acute dosing around the time of injury). In addition, there were dosage issues in these studies, since the dosage of hormone replacement used in many of the clinical studies was supraphysiological (as in the Women's Estrogen for Stoke Trial [WEST]) and noncyclical [147]. This has also been demonstrated in laboratory studies. Cortical explant cultures treated with low doses of 17β-estradiol exhibited significantly reduced cell death while higher concentrations had no protective effects after injury induced by exposure to kainic acid or potassium cyanide/2-deoxyglucose [148].
Estradiol is proinflammatory in the aged brain, most likely secondary to pre-existing damage to the endothelium, and this may also be an important reason for the failure of these trials. 17β-estradiol treatment can lead to enhanced blood–brain barrier breakdown, as measured by Evans Blue dye in reproductively senescent female rats, an effect that is not evident in young estradiol-treated rats [82]. Since several studies have demonstrated a proinflammatory effect of estrogens in diseased vessels [149], it seems that the protective effects of estrogen are limited to the healthy vasculature. The Nurses' Health study exemplifies this issue. In this study, women over 65 years of age were randomized to one of three doses (0.25, 0.5 and 1 mg/day) of 17β-estradiol (E2) or placebo. After 12 weeks of treatment, C-reactive protein (CRP; a general marker of inflammation) decreased 59% in the 0.25 mg/day E2 group and increased 65% in the 1 mg/day E2 group, compared with placebo. The CRP level remained elevated (92%) in the 1 mg/day E2 group, even 12 weeks after treatment was discontinued [150]. In the WHI Observational Study, HRT was associated with higher CRP levels, especially in women who took oral Premarin replacement [151]. Oral Premarin is metabolized by the liver and has numerous effects on clotting mechanisms, producing a hypercoagulable state that probably accounts for the increase in venous thromboembolism observed with treatment. Transdermal formulations of estrogen do not have these adverse effects as systemic routes of drug administration bypass liver metabolism; CRP levels in patients treated with transdermal formulations were similar to untreated women [150,151]. This effect has been seen in laboratory-based murine studies where male mice are more susceptible to thrombus formation than females, possibly owing to sex-specific patterns of hepatic gene expression and effects on coagulation factors [152].
The timing of estrogen therapy is also important. In the WHI trial, women were exposed to Premarin after approximately 10 years of hypoestrogenicity. It has been shown in mice subjected to ovariectomy that 17β-estradiol given shortly after ovariectomy and hormone loss had a significant neuroprotective effect; however, if replacement was delayed by several months, estradiol increased markers of inflammation, such as cytokines and chemokines, and could not salvage tissue from injury [153]. Similar findings have also been observed in primate models, as surgically postmenopausal monkeys fed atherogenic diets had delayed coronary atherosclerosis only if hormone replacement was started immediately after ovariectomy [154,155]. Ongoing studies, including the Kronos Early Estrogen Prevention Study (KEEPS) trial [156] in which women have been randomized to estrogen given orally (conjugated equine) or transdermally (skin patch) within several months of the onset of menopause and followed with carotid intima-media thickness, will hopefully address some of these translational gaps.
The emerging role of testosterone in ischemic sensitivity
The other side of the coin is the male hormone, testosterone. Instead of estradiol mediating intrinsic protection, could testosterone mediate a deleterious effect and be responsible for the ischemic sensitivity in males [157]? Clinically, elevated levels of endogenous testosterone are correlated with an increased risk of stroke in young boys [71,158,159]. Studies in animals have shown that young castrated male rats have decreased injury compared with intact male rats [160]. Testosterone supplementation prior to stroke exacerbated stroke damage in young castrated male rats [157,161,162]. Interestingly, it was also reported that the suppression of testosterone production by a brief anesthetic exposure increased tolerance to ischemia [162]. However, the neurotoxic effects of testosterone are also under debate, as other studies have shown that testosterone can be protective, depending on the age of the animal examined. Testosterone supplementation in middle-aged rats reduced stroke size [160] and enhanced functional recovery when supplemented after stroke in young castrated male rats [163]. Thus, the exact role of testosterone in stroke is controversial.
In humans, testosterone deficiency is seen in approximately 30% of men aged 40–79 years, and, in men with coronary disease, testosterone deficiency is common and impacts negatively on survival [164]. Testosterone replacement in deficient men with such comorbidities ameliorates or partially reverses their progression in some clinical trials [165], but similar to the WHI trial, exogenous testosterone administration increased cardiovascular risk and adverse events in men aged 65 years and older. The excess of cardiovascular events in the testosterone group led the data and safety monitoring board to recommend early termination of the study [166]. Whether these detrimental effects will be seen in younger men (since the average age of men in this trial was 74 years) with fewer vascular risk factors remains to be determined. In addition, it is not known whether these detrimental effects are mediated by testosterone or estrogen (via aromatization). Mice deficient in the androgen receptor display accelerated atherosclerosis, suggesting that testosterone and androgens have their own contributions to vascular health and disease independent of estrogen.

Change in stroke incidence with age and the speculated role of sex hormones.
The role of genetic sex
From the earlier discussion, it is clear that hormones play a critical role in mediating sex differences in stroke. Most of the research to date has focused on the activational effects of acute circulating steroids. These activational effects begin when sex hormones peak at puberty [120]. Therefore, stroke outcomes should be similar in prepubertal children who have equivalent circulating levels of sex hormones [168]. However, prepubertal boys have a higher risk of stroke than girls [159]. In experimental models of neonatal hypoxic injury, activation of sex-specific cell death pathways and sex-specific responsiveness to neuroprotection are seen, despite equivalent hormone levels [76,67]. This suggests that these effects may be due to either the organizational effects of hormones (that occur prenatally and in early postnatal life) or differences in sex chromosomal complement (genetic sex; XX vs XY). Intrinsic genetic sex contributions to ischemic sexual dimorphism have not yet been investigated.
The genetic complement differs in men and women as females have two X chromosomes (partial inactivation of one of the X chromosomes occurs) and males have one X and one Y. The development of testis is secondary to the action of the ‘testis-determining factor’ on the Sry gene. Sry encodes a transcription factor of the high mobility group-box family of DNA-binding proteins that, during embryogenesis, interacts with SF1 (a nuclear receptor) to upregulate Sox9 gene expression [169] prompting Sertoli cell differentiation. A fetus that does not produce functional Sry protein develops as a female (with ovaries) despite having a Y chromosome. Although Sry is chiefly expressed in the testes, it is also expressed in other tissues, including the brain [170].
There are several possible mechanisms through which sex-linked gene expression could contribute to sexual dimorphism in ischemic cell death [171,172]. First, many genes located on the X chromosome (up to 20%) partially escape inactivation [173–177] and could be expressed at higher levels in females. Some X-linked genes are susceptible to progressive escape from X inactivation in an age-dependent manner [178,179] so that sex differences may exist selectively in aged animals. Second, as the two sexes differ with respect to the parental origins of their X chromosomes, any imprinted genes on this chromosome could exhibit sexually dimorphic expression [180–183]. Third, sex differences could be secondary to the male-limited expression of genes in the nonrecombining region of the Y chromosome [184–186] or in the Sry gene itself.
Evidence supporting a role of genetic sex in stroke sensitivity comes from in vitro studies where male (XY) and female (XX) cells or brain slices are evaluated independently of sex steroids [49]. Cortical neurons derived from females (XX) were more sensitive to cell death from stimuli that induce apoptosis compared with XY-derived cells that were more sensitive to nitrosative damage [50]. However, as these cells are taken from late-stage embryonic mice (neurons) or postnatal pups (astrocytes), hormonal effects (organizational) cannot be ruled out in these studies. To specifically dissociate the organizational effects from sex chromosome effects, several investigators have capitalized on the recently developed ‘four core genotype’ mouse model [187,188]. In this model, the Sry gene is deleted from the Y chromosome and inserted on an autosome. Therefore, a cross between a wild-type XX female and a XY male (a male with the Sry deleted from Y but inserted on an autosome, which develops testes) gives rise to four genotypes:
XX males (XX with the Sry on an autosome), which have a ‘female’ chromosome compliment but develop testes due to the presence of the Sry;
XY females (XY ‘male’ chromosomes) lacking the Sry gene, which develop as phenotypic females with ovaries;
XX females (chromosomally and phenotypically female since they have neither the Y nor Sry);
XY males (which have both the Y chromosome and the Sry on an autosome).
A comparison between XY males versus XX males or XX females versus XY females can be used to examine the contribution of sex chromosomes versus gonadal hormones (by comparing XY males vs XY females or XX males vs XX females). Several studies utilizing the FCG mice have successfully demonstrated the importance of sex chromosome complement to pain responses, addiction behavior and neuro-inflammation [189,190]. Stroke studies on these mice should help us dissect out the contribution of genetics and hormones to ischemic stroke sensitivity.
The role of epigenetics
Epigenetic modifications refer to post-trans-lational processes that can modify expression of genes leading to changes in function (often repression of protein synthesis) [191] without any change to the basic DNA structure. These include acetylation and methylation. Acetylation of histone proteins neutralizes their positive charge; decreasing their affinity with the negatively charged phosphates on DNA. This relaxes the condensed chromatin and therefore histone acetylation enhances transcription but deacetylation inhibits transcription [192]. These modifications probably play an important role in underlying sex differences, as a prime example of epigenetic regulation comes from the sex chromosomes themselves. Random inactivation of one of the X chromosomes is mediated by epigenetic modifications [193]. X-inactive specific transcript (Xist) RNA expression, methylation of CpG islands and hypoacetylation of histone H4 have now been established to be important features of the inactive X chromatin [194]. However, many X chromosome genes escape inactivation. It is largely unknown as to how these processes are regulated and which genes are targets of inactivation, but will probably be a major focus of research in the future for several reasons. Histone deacetylase inhibitors have been recently demonstrated to be neuroprotective after stroke suggesting that these will be a target of drug development [195]. Since sex differences in levels of histone modification have already been described in the cortex and hippocampus of neonatal mouse brains [196], future studies will have to test the efficacy of ‘epigenetic therapies’ in both the sexes.
Recent reports indicate that prenatal testosterone treatment increases acetylation and subsequent gene transcription in neonatal female cortex/hippocampus (which normally have low histone acetylation levels) [196]. This provides an example of not only the organizational effects of hormones that may not manifest themselves until later in development, but also of the epigenetic modifications caused by prenatal secretion of sex hormones. However, these effects appear to be region dependent, as the preoptic area, hypothalamus and amygdala do not show sexual dimorphism in histone acetylation [196]. In addition to the sex hormones themselves, it has been shown that their receptors (estrogen and androgen receptors) are acetylated and methylated to varying degrees [197–199]. Estrogen receptor (ER)a, due to its role in mediating the acute neuroprotective effects of estrogen, is of particular interest [130]. The expression of ERa mRNA is known to be high early in the postnatal period in the cortex, but its levels decline throughout development [200]. This is owing to methylation-induced gene silencing in the brain leading to low levels of transcription. Cortical ERa 5' exons A and C are methylated at time points that correspond to the significant decrease in ERa mRNA expression. Importantly, it has recently been shown that this methylation is a dynamic process that is emerging as an important mechanism by which estrogen signaling can be enhanced after injury [198]. Cortical ERa gene expression increases rapidly after stroke in females [201], and this enhanced expression is thought to be responsible for the neuroprotective effects of estradiol in stroke [202]. This increase in the ERa expression is a result of the decrease in methylation of the promoter allowing for transcription of estrogen responsive genes [203], some of which, like BDNF, are known to enhance neuronal survival and repair [204]. Interestingly, it was found that post-MCAO, MeCP2 (the methyl-DNA binding protein) was dissociated from the ERa promoter only in female rats, which corresponds with the methylation status of the promoter. This suggests that ERa expression is regulated differently in males and females in response to MCAO, indicating that the brain can revert back to a developmental program of gene expression after stroke, a topic of a recent review [205].
Although the expression levels of ERa decrease similarly with development in the cortex and hippocampus of both males and females [206,207], other areas of the brain, such as the hypothalamus, are rich in estrogen receptors and show sex differences in expression [208]. The medial preoptic area shows higher expression of estrogen receptors compared with males [208,209]. Thus, ERa expression in the preoptic area of the hypothalamus becomes sexually dichotomized during development [209–211] and appears to remain so during adulthood [212–214]. This sexual dimorphism in the expression of ERa receptors is important since these receptors play an important role in organizing sexual behaviors in males and females [209,215]. Hence, the expression of ERa receptors is regionally and temporally [205] regulated. Paradoxically, methylation in the ERa promoter of neonatal females is higher, which would be predicted to lead to reduced expression and not the higher mRNA and protein levels as observed [210,211]. Therefore, not all the epigenetic changes are linked directly to gene expression.
Differences in men and women in stroke risk are also emerging. The Oxford vascular studies reported that women were approximately 50% more likely to have a maternal history of stroke, while no such risk exists in males [216]. Although this may be explained by mitochondrial gene transmission, it appears that both genetic and epigenetic factors may contribute to the higher maternal transmission of risk to daughters. Since behavioral programming and environmental factors can alter the epigenomic state of a gene [217], it may be possible that this observation in the Oxford study is attributable to the fact that mother and daughter share the same socioeconomic (environmental) risk factors for stroke. The ‘Barker Hypothesis’ stated that low birth weight/suboptimal environment in utero leads to an increased vascular disease risk [217,218]. Thus, it appears that in utero environment has its own important role to play in later stroke risk.
Admittedly, our knowledge of epigenetic changes in the developing and mature brain is still in its infancy. A thorough understanding of the post-translational modifications and the orchestrated interplay with injury, development and changing levels of sex hormones throughout the lifespan is needed to further the study of sex differences.
Future perspective
The available clinical and basic science literature provides strong evidence for sexual dimorphism in ischemic stroke. Although adult females have an intrinsic ‘ischemia-protected’ phenotype after experimental stroke, much of this is secondary to gonadal hormones. Future research will need to incorporate aging models into experimental stroke studies. Although estradiol has been proved to be a neuroprotective agent in many animal studies, it has failed as a therapeutic agent in clinical trials. Ongoing clinical studies have addressed some of these issues, by initiating estrogen replacement at the time of menopause, and administering transdermal formulations that avoid first pass metabolism through the liver. Hopefully, results of the Kronos Early Estrogen Prevention Study (KEEPS; expected in 2012) will clarify the role of estrogen in stroke risk. Since most of the preclinical evidence suggests that estrogen is a potential neuroprotective agent in many animal models, pilot trials for acute use in stroke patients should also be considered. As estrogen is currently in clinical trials for treatment of acute traumatic brain injury, if therapy is safe this therapy could be extended to stroke patients. Preclinical studies showing that estrogen can enhance stroke-induced neurogenesis [89,219] may also be of relevance for the development of future therapeutic trials. It will be increasingly important to examine the long-term effects of estradiol in clinical trials on chronic functional recovery after stroke. Studies on the role of androgens on ischemic risk are also needed.
As the sensitivity of each sex to specific cell death pathways acts independently of adult hormone levels, factors beyond the activational effects of hormones contribute to ischemic sexual dimorphism. The contribution of sex chromosome dosage, early prenatal organizational effects of hormones, and epigenetic factors, or the interaction between these, promises to be a complex but rewarding area for future researchers in both the clinic and in the laboratory. Importantly, the underlying molecular mechanisms that contribute to sex differences in ischemic cell death must be further elucidated. Even if the degree of ischemic injury is equivalent between the sexes (i.e., in a young male and a recently ovariectomized female), if cell death is triggered by different, sex-specific mechanisms it is likely that different therapies will need to be developed for each sex.
Executive summary

Males have a higher incidence of stroke compared with females until the age of 85 years when the incidence reverses.
Women bear the major burden of stroke-related disability and institutionalization.
The incidence of a recurrent stroke is higher in women versus men.
There are many in vitro and in vivo models in the laboratory that mimic ischemic stroke.
Stroke patients differ in their age, sex and comorbidities and our models need to incorporate these differences to enhance translational relevance.
In vivo models of ischemic stroke have demonstrated that young females have smaller infarcts as compared with young males after an equivalent insult.
Aging female rodents lose the intrinsic female protection ‘phenotype’ that is observed in young females and have larger infarcts as compared with aging males.
In vitro models of stroke using oxygen glucose deprivation and NMDA toxicity have demonstrated that female-derived neuronal cultures or brain slices derived from females have improved survival after injury compared with males.
Ischemia-induced cell death pathways differ between the two sexes, which determines the response to neuroprotective agents.
The caspase-independent (nitric oxide/poly[ADP-ribose] polymerase) cell death pathway predominates in males.
The loss of poly(ADP-ribose), nitric oxide synthase or apoptosis-inducing factor deficiency reduces infarct in males, but has no effect or paradoxically increases infarct in females.
Similarly, nitric oxide synthase knockout males have smaller damage versus females, which have increased injury.
Caspases are mediators of cell death in females.
Pan caspase inhibitors decrease infarct volumes in females but have no effect in males.
The etiology of the sex differences seen in ischemic stroke both clinically and in experimental models is multifaceted.
Hormones have organizational and activational effects.
Estradiol is recognized as a neuroprotectant.
In rodents, ovariectomy increases infarct size in females and estradiol supplementation reduces injury.
Clinical studies of chronic estradiol replacement showed an increase in stroke incidence in treated postmenopausal women.
The role of testosterone is still a subject of debate and effects may depend on the age of the patient or animal examined.
Genetic sex may play an important but unknown role in the etiology of sex differences in stroke.
Epigenetic modifications can modify expression of genes and their functions by acetylation or methylation and this post-translational modification may be an important etiology of the sex differences in stroke.
The mechanisms underlying the ‘Barker effect’ that low birthweight/suboptimal environment in utero leads to an increased vascular disease risk remains to be investigated in experimental models of stroke.
The Oxford vascular studies suggesting that a maternal history of stroke is important to adult stroke risk in female offspring needs to be modeled and investigated.
Future studies need to be carried out in order to dissociate the role of genetic sex from hormones.
Current literature has primarily used young male animals to model stroke.
Approximately three quarters of stroke patients are aged over 65 years.
Different physiological and inflammatory responses occur in the aged brain after injury.
The Nurses' Health Study made it clear that estradiol can induce a proinflammatory effect by increasing the C-reactive protein levels.
Therapies efficacious in the young may not be effective in the aging population.
Elderly women bear the major brunt of ischemic stroke in terms of poor functional outcomes and increased stroke severity.
Very few studies have examined aging female animal models and even less is known regarding chronic recovery after injury.
The ‘four core genotype’ model will be a useful tool to uncouple the effects of genetic sex from hormones in the etiology of sex differences in stroke.
Footnotes
This work was supported by a NIH grant: NIH R21 NSO66406 (to Louise D McCullough). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.