
Abstract
Cardiovascular disease (CVD) is the number one cause of morbidity and mortality in men and women worldwide. According to the WHO, by 2015, almost 20 million people will die from CVD each year. It is well established that men and women differ not only in baseline cardiac parameters, but also in the clinical presentation, diagnosis and treatment outcomes of CVD. Women tend to develop heart disease later in life than men. This difference has been attributed to the loss of estrogen during the menopausal transition; however, the biological explanations for the sexual dimorphism in CVD are more complex and seem unlikely to be due to estrogen alone. The current controversy that has arisen regarding the effects of HRT on CVD in women is a case in point. In this review, the sex-based differences in cardiac (patho-) physiology are discussed with emphasis on the impact of sex hormones, hormone receptors and diet on heart disease.
Keywords
Biological sex plays an important role in cardiac pathophysiology. The incidence of heart disease in women increases after menopause, indicating that sex hormones play a critical role in the development of heart disease. HRT and dietary interventations are commonly undertaken for the prevention and management of cardiovascular disease (CVD). In this review, we highlight the fundamental differences in cardiac function between men and women. The emphasis is on the role of sex hormones and phytoestrogens on heart disease. By drawing on a breadth of data from studies in humans as well as animal models, it is possible to identify important effects of the role of sex hormones and phytoestrogens on heart disease.
Sex differences in heart function
Sex-based differences in normal heart function have long been recognized; however, in more recent years the influence of sex on heart disease has been explored at cellular, molecular and genetic levels. Before puberty, the left ventricular (LV) mass normalized to body weight is only modestly higher in boys than in girls (mean of ∼6%) [1]. In all older age strata, the LV:bodyweight ratio in men is 25–38% greater than in women [1,2]. Given that the number of human cardiac myocytes is determined within the first year of birth, when the mitotic activity of normal myocytes appears to cease, any subsequent increase in heart size is primarily due to changes in myocyte size (hypertrophy) rather than in their numbers [3]. A larger LV mass in men reflects a greater degree of hypertrophy compared with women. This LV hypertrophy has been demonstrated to be due to a symmetrical increase both in chamber dimension and in wall thickness in males, resulting in no sex difference in relative wall thickness [1].
Sex also plays a critical role in the detrimental effects of the aging process in the heart. An inter-esting observation in this context is the preservation of myocardial mass, myocyte number and average cell diameter and volume in women through the ages of 20–95 years. By contrast, men between the ages of 17–89 years demonstrate a gradual loss of nearly 1 g/year of the myocardium, accounting for approximately 64 million cells lost from both the left and right ventricles [4,5]. In the remaining cells, myocyte cell volume increases (hypertrophy) at a rate of 158 μm3/year in the left and 167 μm3/year in the right ventricle in males [5]. The underlying mechanism(s) for this difference between male and female hearts remains to be elucidated. A previous report using hearts from human subjects ranging from 21 to 93 years of age demonstrated that there is no correlation between age and cardiomyocyte apoptosis between the two sexes; however, in this study, cardiomyocyte apoptosis was threefold higher in men compared with women in all age groups [4]. The fact that apoptosis and age are not correlated does not rule out the possibility of other forms of cell death (i.e., necrosis) occurring in aging cardiomyocytes. The sex difference in apoptosis could in part be explained by the role of estrogen in protecting cardiomyocytes from apoptosis in women.
Calcium handling
In general, the force of contraction in a normal myocardium is comparable between males and females [10–12]. However, the durations of contraction and relaxation are usually shorter in the female myocardium, and the maximum velocities of tension development and decline are also markedly faster in the female myocardium [10,13]. An overt sex-related difference also exists in myofilament Ca2+-responsiveness that is believed to contribute to the disparity of myocardial contractile function between men and women. The female myocardium seems to possess a lower myofilament Ca2+-sensitivity than the male myocardium [13]. Myocellular Ca2+-handling is the key determinant of normal cardiac contractile function. Perturbations in Ca2+-handling lead to cardiac dysfunction and HF [14–16] characterized by attenuation in force development and impaired relaxation in the cardiac muscle [17]. Functional changes in contractility are associated with changes in the expression of key Ca2+-handling proteins [18,19]. For example, alterations in the expression of myocellular proteins such as the sodium–calcium exchanger (NCX) [20–22] and sarcoplasmic reticulum calcium ATPase (SERCA) have been well-documented, both in clinical HF and experimental models of HF [23–26]. Sex-dependent differences in the expression of these proteins exist in normal and failing myocardiums, and may play a role in how the heart responds to cardiac dysfunction in both sexes. NCX protein levels in humans are consistently higher in female hearts compared with male hearts [22]. However, previous reports indicate that in both male and female failing hearts, NCX levels are higher than in nonfailing hearts; however, this increase is proportionately greater in males [20–22]. By contrast, SERCA protein levels are downregulated in human HF in both males and females [27]. However, reports from an animal model of pressure overload hypertrophy have demonstrated that
Sex differences in the manifestation of cardiovascular disease
Cardiovascular disease manifests itself differently in women compared with men. Several scenarios in which sex plays a critical role in this context in humans and in animal models are discussed in this section. Table 1 describes sex differences in the manifestation of CVD in various genetically manipulated mouse models.
Differences in the manifestation of cardiovascular disease in male and female mice.
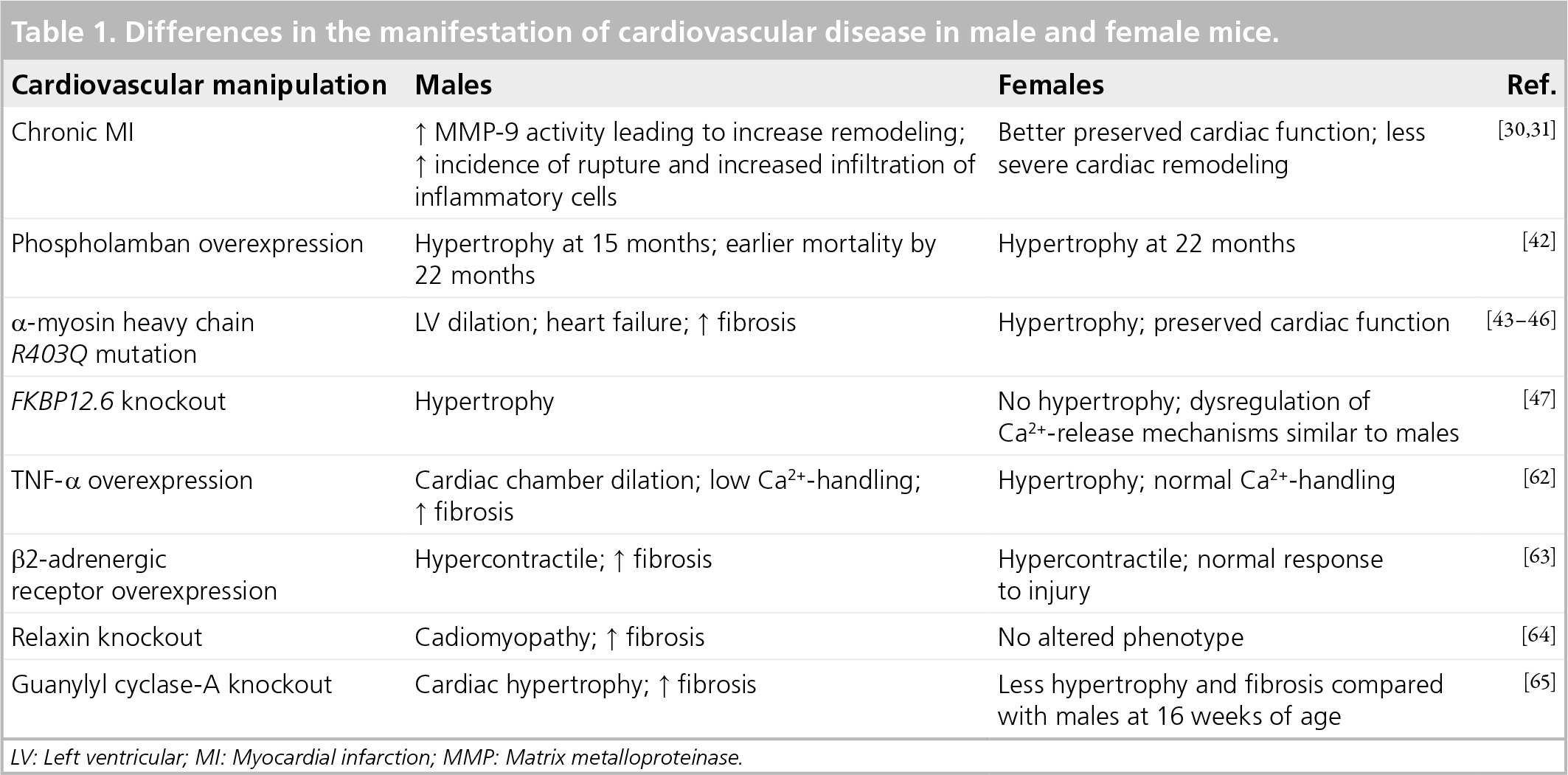
LV: Left ventricular; MI: Myocardial infarction; MMP: Matrix metalloproteinase.
Myocardial infarction
In the USA, the median age for the first MI is 65.8 years for men and 70.4 years for women [29]. Women are more likely than men to die as a result of a MI; 38% of women die within 1 year of their first MI compared with 25% of men [29]. This could be attributed to the fact that women are more likely to be older when they have their first MI compared with men [29,30]. In addition, women are more likely to be misdiagnosed after their first MI because of their atypical symptoms [31]. Perimenopausal women rarely experience chest pain as typically expected, making a proper diagnosis more difficult. Chest pain, described by men, which often precedes MI, does not necessarily indicate MI in women. Angina in women is only predictive of coronary heart disease (CHD) in 50–60% of cases, whereas it is predictive of CHD in 80–99% of cases among men [31]. Factors that indicate MI in women with more effectiveness than in men include shoulder pain, neck pain, dyspnea and fatigue [31]. Studies using mice after a chronic MI indicate that females have a better preserved cardiac function following MI; in addition, the LV remodeling in females is less severe compared with their male counterparts [32,33]. The cardiac rupture rate following MI is significantly lower in females compared with males, consistent with less remodeling in female hearts [32–34]. This sex-specific dimorphism in response to MI could also be attributed to the increased activity of matrix metalloproteinases (MMP-9) leading to an enhanced degradation of extracellular matrix proteins in male mice [35]. In addition, hearts from female mice subjected to an MI were demonstrated to have markedly less infiltration of inflammatory cells compared with males, which could also contribute to the lower incidence of rupture in females [34]. However, the literature on cardiac rupture in humans remains controversial. Shapira
Myocardial hypertrophy
Hypertrophy
Arrhythmias
Sex-based differences in cardiac electrophysiology play a crucial role in the prevalence of different clinical arrhythmias. In general, women have faster resting heart rates (3–5 beats/min faster) [50] and rate-corrected QT intervals compared with men [51]. In addition, sex differences in T-wave shape have been observed [52]. The interval from S-wave offset to the peak of T-wave is also significantly prolonged in women compared with men [53]. The action potential duration in women is longer at all of the phases of an action potential compared with men [52]. Atrial fibrillation (AF) is the most common arrhythmia observed in clinical practice today, with an incidence rate that increases with age [54]. In the Framingham Heart Study, men were found to have a 1.5-fold higher risk for developing AF compared with women [55]. However, the mortality rate owing to AF is similar between men and women. Therefore, AF diminishes women's survival advantage that is normally observed in other forms of heart disease [56]. Furthermore, women with AF are more likely than men to have embolic strokes [57]. Female gender is also an independent risk factor for both congenital and acquired long QT syndrome [54,58]. The incidence of syncope and sudden death associated with congenital long QT syndrome is also higher in women. A major threat attributed to the prolongation of QT interval is the life-threatening Torsades de Pointes (TDP), which is usually induced by antiarrhythmic drugs and is also more common in women than men [59]. The mechanisms underlying these sex differences with respect to arrhythmia are not completely understood; however, sex hormones are thought to be involved. A similar propensity towards drug-induced TDP was observed in both pre- and post-menopausal women, suggesting that estrogen does not have a protective role [60]. In addition, women taking oral contraceptives reportedly have a higher incidence of ventricular arrhythmia, suggesting that estrogen or progesterone could be arrhythmogenic [61]. It has been suggested that the actual ratio of estrogen to progesterone, rather than the absolute levels of the two hormones, might determine the propensity for women to develop TDP [62,63].
Cardiac fibrosis
Cardiac fibrosis is a hallmark of various forms of heart disease. It is characterized by an enhanced deposition of extracellular matrix proteins – most importantly collagen – leading to an impairment of diastolic and systolic function. Fibrosis develops following myocyte death, inflammation, pressure and volume overload, hypertrophy and stimulation by a variety of hormones, growth factors and cytokines. Sex differences in the progression of fibrosis have been postulated clinically. Biopsies from patients with aortic stenosis have demonstrated an increased myocardial stiffness, more common endocardial fibrosis and abnormal collagen architecture in males compared with females. Similarly, in various animal models of HF, more severe cardiac fibrosis is observed in male mice compared with females. For example, transgenic mice with overexpression of TNF-α [64], a mutant α-MHC [45,46] or β2-adrenergic receptor [65], and relaxin (a peptide hormone) [66] null mice present with increased fibrosis in males. Guanylyl cyclase-A (GC-A) knockout mice also exhibit fibrosis, which is significantly more pronounced in male mice compared with females [67].
Differential response of the two sexes to exercise
Exercise is an important factor in preventing CVD. Studies suggest that the favorable physiological responses to exercise might slow some of the pathophysiological progression of HF [68,69]. Exercise is known to activate complex interactions of central hemodynamics, peripheral blood flow, skeletal muscle, endothelium, neurohormones and the cytokine system, all of which benefit patients with HF [70–75]. The types, intensity, duration and frequency of exercise that could have positive effects on HF outcomes have still not been clarified. The role that sex plays in cardiac adaptation to exercise is also unclear. A sex-based dichotomy exists with respect to cardiac adaptation to exercise. Women tend to exhibit a relatively smaller increase in LV ejection fraction in response to exercise compared with men. One explanation for this differential ventricular response may be related to the different mechanisms by which women and men increase cardiac output during exercise. Women tend to increase their cardiac output primarily by increasing end-diastolic volume index without significantly increasing ejection fraction; this is compared with men who primarily decrease end-systolic volume index and raise ejection fraction [76]. Data from mice also suggest that exercise provokes a sex-dependent cardiac adaptation. Female mice have an increased voluntary and forced exercise capacity and increased hypertrophic response to similar amounts of exercise [77]. Chronic exercise significantly decreases the ratio of the slower β-myosin isoform relative to the α-myosin isoform in female mice while males experience no change. Taken together, these data suggest that females tend to benefit more from exercise than males. Several signaling pathways are implicated in affecting cardiac adaptation to exercise. Potential candidates for exercise-mediated cardiac hypertrophy are
Differential response of the two sexes to cardiovascular therapeutics
Women have been under-represented in the majority of the cardiovascular clinical trials in the past, which has left many questions unanswered in interpreting the efficacy of various drugs in women. There is evidence suggesting the existence of sex-specific differences in clearance rates, bioavailability and the efficacy obtained with the same dose of the drugs used for treating CVD [81]. Primary drug-metabolizing enzymes in the intestinal wall and liver, which are part of the cytochrome P450 family, have different activities in men and women [81]. Recent data regarding the efficacy of digoxin therapy, which is independent of age, suggest that it is associated with an increased mortality in women, but not men, and that is also associated with HF and depressed LV systolic function in women [82]. Although the digoxin dosage was standardized to BMI, women demonstrated slightly higher serum levels of the drug compared with men, indicating a difference in pharmacokinetics between the sexes [82]. A systematic meta-analysis including 15 trials indicates that statins reduce the risk of cardiovascular events in both men and women; however, women on statins may not have the reductions in mortality, MI and stroke that are observed in their male counterparts [83]. In general, women have higher serum levels of statin and the risk of adverse drug reactions owing to statins is relatively higher in women [84]. Women have a faster clearance rate, and hence lower serum levels, of certain Ca2+-channel blockers (e.g., verapamil and nifedipine) than men. Reduction in blood pressure owing to Ca2+-channel blockers is more pronounced in women [81]. Adverse drug reactions in the form of coughs while being treated with angiotensin-converting enzyme (ACE) inhibitors occur twice as frequently in women [85]. It is therefore very important to study cardiovascular drugs in both sexes.
Sex steroids & sex steroid hormone receptors in the heart
Sex steroids
Men and women both possess significant levels of estrogen and testosterone. In women, estrogen is synthesized primarily in the ovaries and, to a lesser extent, in the pituitary gland, adipose tissue and the liver, while testosterone is synthesized in the ovaries and in the adrenals. In men, estrogen is synthesized in the testes, the pituitary gland, the liver and in adipose tissue, while testosterone is synthesized mainly in the testes and, to a lesser extent, in the adrenals.
Estrogen has been suggested to be cardioprotective in many different settings [86–88]. There have been many proposed mechanisms for the cardioprotective effects of estrogen. First, estrogen has been demonstrated to induce mitochondrial biogenesis [89]. Second, many cardioprotective genes are upregulated either directly or indirectly by estrogen, including PGC-1α [90], MCIP1 [91] and HSP72 [92]. Estrogen also activates nitric oxide synthase and Akt through the PI3 kinase pathway, which has been shown to confer protective effects on both cardiomyocytes and endothelial cells [93,94]. Finally, estrogen has been shown to upregulate the production of cardiac natriuretic hormones, which have several beneficial effects on cardiac function, including reducing blood pressure and improving endothelial function [95]. Testosterone has been far less studied and so little is known about its underlying mechanism(s) of action in the heart. Testosterone has been demonstrated to induce hypertrophy and fibrosis in the murine heart [67]. There is evidence suggesting that the prohypertrophic effect of testosterone is mediated through the mTOR axis [96]. Testosterone has also been shown to be anti-inflammatory [97] and is involved in regulating antioxidant levels [98]. Furthermore, testosterone has been demonstrated to be important in maintaining the normal vascular tone in males through its conversion to estrogen in the vasculature by the enzyme aromatase [99].
Sex steroid hormone receptors
The biological effects of sex steroids are mediated by sex steroid hormone receptors (SSHRs). Sex steroids act as ligands and activate their cognate receptors; estrogen activates ERs (i.e., ERα and ERβ) while androgens activate androgen receptors (ARs). Upon ligand binding, the SSHRs dimerize and bind to specific DNA response elements, and engage the general transcriptional apparatus. These are defined as the genomic actions of SSHRs. SSHRs also elicit their effects in the target tissues by recruiting secondary messengers and kinase signaling cascades [100,101]. These responses occur too rapidly to involve changes in target-gene transcription and are therefore regarded as nongenomic [102]. Both of these modes of action of SSHRs have been previously reviewed in detail [100–104]. Most research is focused on the actions of ERs in cardiac pathophysiology but very little is known regarding ARs. In fact, all the SSHRs are expressed in both male and female cardiac myocytes, suggesting their importance in cardiac physiology. Levels of ERα (both mRNA and protein) are equivalent in the hearts of both men and women. Levels of ERβ mRNA, on the other hand, are higher in male than in female hearts [105]. Both ERα and ERβ are upregulated during human aortic stenosis [106]. Similarly, myocardium from patients with endstage HF demonstrates elevated expression of ERa mRNA [105]. AR mRNA is expressed in cardiac tissues from both men and women [107]. Rat myocytes respond to androgens by eliciting a hypertrophic response [107]. Hence functional SSHRs are expressed in the heart and play important roles in cardiac pathophysiology. A better understanding of these receptors in the heart could elucidate the sexual dichotomy observed in cardiac function to a considerable extent.
Genetic studies on ER & AR
The receptors ERα, ERβ and AR are expressed at equivalent levels in male and female mice hearts [108]. ERα, ERβ and AR knockout mice have been used extensively to investigate the effects of hormones on pathological cardiac remodeling. The hearts of ERα knockout (αERKO) mice do not exhibit any baseline cardiovascular differences [109]. However, after short-term ischemia-reperfusion (I/R), αERKO females display reduced function and increased apoptotic signaling compared with wild-type controls. Interestingly, αERKO males do not exhibit a differential phenotype compared with wild-type mice post-I/R [110]. Similarly, compared with wild-type controls, female βERKO mice have increased expression of caspase 3 and 8, two proapoptotic markers, post-I/R. Interestingly, male βERKO knockout mice display decreased apoptotic signaling post-I/R compared to wildtype controls, implying a sex-dependent function of ERβ [111]. The available data suggest that both ERα and ERβ are protective in females, but not males, after short-term I/R.
By contrast with αERKO mice, both male and female βERKO mice display hypertension and increased heart mass prior to cardiovascular manipulation [112]. βERKO female mice subjected to ovariectomy followed by estrogen supplementation display an increased expression of atrial natriuretic peptide and increased mortality compared with wild-type controls post-MI [113]. By contrast, another study using the same methods as described previously reported reduced mortality in βERKO females post-MI compared with wild-type controls [114].
Endoplasmic reticulum-α knockout and βERKO mice have also been used to investigate the hormonal effects on pathological cardiac hypertrophy in response to pressure overload. In response to transverse aortic constriction (TAC), which induces pressure overload, estrogen attenuates the development of cardiac hypertrophy [115]. However, this protective antihypertrophic effect of estrogen is abolished in female βERKO mice, but not in female αERKO mice, suggesting that ERβ is responsible for the antihypertrophic properties of estrogen. Interestingly, this effect was not observed in male βERKO mice [114,116].
Male AR-knockout (ARKO) mice display reduced baseline heart weight and reduced myocyte cross-sectional area, suggesting a prohyper-trophic effect of testosterone [117]. Interestingly, after angiotensin II stimulation, ARKO males display increased arterial medial thickness and perivascular fibrosis compared with wild-type controls, implying an important protective role for testosterone in the vasculature [118]. The GC-A knockout (
The majority of the experimental data point to ERα and ERβ playing a protective role, especially in females; however, this hypothesis is far from proven. Importantly, all of the previous studies were on global nulls of ERs. Studies using conditional cardiac-specific knockouts would help to clarify whether ER activity is directly beneficial or detrimental to cardiac function. More could be learned regarding the cardiovascular effects of estrogen from the aromatase-knockout mouse. These mice are lacking an essential enzyme for synthesizing estrogen. The aromatase-knockout mice are reported to have a 5% lower blood pressure and a 46% lower baroreflex sensitivity compared with wild-type controls [119].
Gonadectomy & hormone replacement in mice
Gonadectomy and hormone replacement can also alter the cardiac response to pathological stimuli (Table 2). Ovariectomized females subjected to volume overload develop greater cardiac hypertrophy, dilation and pulmonary edema than intact females [86]. Ovariectomized mice treated with a high physiological dosage of estrogen and subjected to pressure overload develop less severe cardiac remodeling than placebo treated mice [87]. Patten
The cardiovascular effects of gonadectomy and hormone supplementation in mice.

MI Myocardial infarction
There are several caveats to consider when interpreting data in the literature. The effects of hormone replacement or removal experiments on the heart depend greatly on the stimulus, duration and the dose of hormone treatment, and the sex of the mice. Another important caveat is that measured hormone levels in intact wild-type mice vary dramatically depending on the laboratory and the method employed. Measurements of estrogen levels in adult female mice reported in the literature vary from 2.9 to 99 pg/ml [113,121]. By contrast, testosterone levels in adult male mice vary from 35 to 1500 ng/dl [122,123]. Therefore, it is essential that any study reporting hormone supplementation at a physiological level must measure wild-type mice, gonadectomized mice treated with placebo and gonadectomized mice supplemented with hormones. As a final caveat, most studies have focused on the effects of hormones on cardiomyocytes, and it is important to consider that the heart consists of a myriad of cell types besides cardiomyocytes, including cardiac fibroblasts and endothelial cells. The heart is 90% myocyte by mass, but by cell number, cardiomyocytes make up only 25–35% of the total number of cells. Cardiac fibroblasts constitute 90% of nonmyocyte cells [124]. Recently, the presumption that apoptosis in the myocardium primarily occurs in myocytes was challenged by a study using a monkey model of HF. This study found that the majority of apoptosis occurred in nonmyocytes [125]. Nonmyocyte cell types in the heart, such as endothelial cells, smooth muscle cells and fibroblasts, all express ERα, ERβ and AR. Therefore, the hormonal effects of estrogen and testosterone on nonmyocytes should also be considered when drawing experimental conclusions [126,127]. It should also be noted that results obtained using rodent models may not necessarily be extrapolated to humans owing to differences in cardiovascular biology, such as heart rate and molecular composition.
HRT in women
As mentioned previously, incidence of heart disease is sexually dimorphic in men and women younger than 65 years of age (CDC). However, when comparing men and women over 65 years of age, this sexual dimorphism disappears and mortality is higher in postmenopausal women compared with age-matched men [201]. This led to the hypothesis that estrogen is a cardioprotective hormone in women and that the reduction in estrogen levels in postmenopausal women causes this increase in the incidence of heart disease.
Many observational and randomized clinical trials have been conducted to investigate the relationship between menopause with or without HRT and heart disease. Observational studies have provided evidence that menopause is associated with an increased risk of heart disease. Age-matched women who undergo natural menopause prior to 50 years of age have a 1.5-fold higher rate of CHD than those who undergo menopause after 50 years [128]. In age-matched women who undergo surgical menopause, the rate of CHD is approximately twofold higher than hormonally intact women [129]. Based on these studies, postmenopausal women were prescribed estrogen for the primary purpose of preventing heart disease. A meta-analysis of 25 observational studies demonstrated a decreased risk of CHD with estrogen treatment alone. The hazard ratio, defined as the ratio of CHD occurrence of hormone users to nonhormone users, was 0.70. By contrast, in women receiving estrogen plus progesterone treatment, the hazard ratio was 0.66 [130]. However, it is important to note that these trials were not randomized, which may be a confounding factor.
The randomized Heart and Estrogen/Progestin Replacement Study (HERS) trial was designed to study the effects of hormone replacement in postmenopausal women with established coronary disease and an intact uterus. In the HERS trial, 2763 postmenopausal women were given either 0.625 mg/day of conjugated equine estrogen (CEE) plus 2.5 mg/day of medroxyprogesterone acetate (MPA) or a placebo. Women who received CEE plus MPA did not show a reduced occurrence of MI or CHD death with a hazard ratio of 0.99 [131]. Compared with placebo, hormone treatment was not beneficial in women with established CHD.
The Women's Health Initiative (WHI) involved 16,000 women and was the first major randomized trial to test the hypothesis that replacing the endogenous estrogen lost owing to menopause with exogenous estrogen would protect women from developing heart disease. The results from the WHI study proved less conclusive than previous observational studies.
In women with an intact uterus receiving CEE plus MPA, hormonal treatment increased CHD risk with a hazard ratio of 1.29 [132]. In women with prior hysterectomy treated with CEE alone, hormone treatment did not change CHD risk [133]. Secondary analysis of data from the WHI trial suggested that initiating hormone treatment soon after menopause might reduce CHD events, with a nonsignificant hazard ratio of 0.76 for women who started hormone replacement within 10 years of menopause compared with women who started hormone replacement more than 10 years postmenopause [134]. The idea that initiating hormone treatment soon after menopause might protect from CHD is termed the timing hypothesis. However, further analysis of the WHI trial, including data from both observational and randomized trials, did not demonstrate a reduction in CHD events in women who started hormonal therapy closer to menopause [135]. Thus, the cardiac benefits of HRT remain unresolved. Moreover, it should be noted that in the CEE plus MPA arm of the WHI trial, HRT significantly increased the risk of breast cancer, stroke and pulmonary embolism, and caused the trial to be stopped early. On the other hand, HRT reduces some risk factors for CVD. One such risk factor for which prevalence is reduced by HRT is central obesity, a strong predictor of CVD occurrence [136,137].
Phytoestrogens & cardiovascular health
Given the risks associated with conventional HRT, there is growing scientific interest focused on alternative therapies for postmenopausal women with a predisposition to CVD. Phytoestrogens are interesting candidates in this regard since they are structurally similar to estrogens (Figure 1). They act as both estrogenic agonists and antagonists [186].

Structural similarities between 17β-estradiol and phytoestrogens.
The phytoestrogens daidzein and genistein are naturally occurring isoflavones that are found in numerous edible plants, especially soybeans. Individuals consuming soy on a regular basis (∼50 mg/day) could have plasma isoflavone concentrations of 50–800 ng/ml, which is 100-times higher than endogenous estradiol levels (concentrations in men and women ranging from 40 to 80 pg/ml) [138]. In infants fed on soy formula, these concentrations are even higher, reaching 9–10 μM [138]. Phytoestrogens bind both ERα and ERβ, but they bind ERβ with greater affinity (Table 3) [139]. The relative potency of phytoestrogens in activating transcription of estrogen-responsive genes is 1000- to 10,000-fold less compared with 17β-estradiol, indicating that by comparison with 17β-estradiol, genistein and daidzein have weak genomic estrogen effects (Table 4) [140].
Relative estrogen potencies of 17β-estradiol and phytoestrogens.

The table illustrates the relative genomic estrogenic activity of the phytoestrogens in comparison with 17β-estradiol, arbitrarily set at 100.00.
Adapted from [184].
Relative binding affinities of 17β-estradiol and phytoestrogens for estrogen receptors α and β.

The relative binding affinity of 17β-estradiol was arbitrarily set at 100.00. Adapted from [185].
Isoflavones in soy foods and supplements occur as highly water-soluble glucosides, which undergo hydrolysis by intestinal β-glucosidases to release bioactive aglycons (i.e., daidzein and genistein) prior to absorption by the small and large intestines. After absorption, the isoflavone aglycons are reconjugated with glucuronic acid and, to a smaller extent, with sulfate by the phase II enzymes UDP- glucuronosyltransferases and sulfotransferases in the liver. Similar to endogenous estrogens, these conjugates are excreted via both urine and bile. Only 7–30% of the ingested amount of daidzein and genistein can be recovered in urine and less than 10% in the feces [141,142]. This low recovery could be explained by further metabolism of both genistein and daidzein by the hepatic cytochrome p450. Daidzein undergoes further metabolism by means of the gut microflora [143] to obtain a biologically active metabolite, equol, which is more estrogenic than daidzein (Table 4) [144]. Equol also has higher affinity for both ERa and ERβ than its precursor daidzein (Table 3) [145,146]. Among humans, approximately 30–50% of the population are equol-producers. By contrast, mice, rats, hamsters, cows, pigs, sheep, dogs, monkeys and chimpanzees all have the ability to produce equol [147–151]. Diet and host genetics may contribute to inter-individual variations in equol production in humans. However, the reasons for such differences in harboring the equol-producing bacteria remain essentially unknown. The specific bateria or bacterium responsible for equol production in the human gut is also yet to be identified. According to Setchell
The clinical effectiveness of soy protein in cardiovascular health is equivocal. In 2006, the American Heart Association reversed its endorsement of soy products by stating that it found no effect of soy proteins or plant estrogens on blood pressure, or menopausal changes [154]. By contrast, other human studies indicate that soy could have potential beneficial impacts on cardiovascular risk factors, including body mass index [155], lipid profiles [156,157], glucose and insulin homeostasis [158,159] and hypertension [157]. In the majority of 22 randomized trials, isolated soy protein decreased LDL-cholesterol concentrations by an average effect of approximately 3%. This reduction is very small relative to the large amount of soy protein tested in these studies, averaging 50 g, which is approximately half the usual total daily protein intake [160]. Isoflavones have been shown to induce vasodilation by stimulating the activity of the endothelial NO synthase (NOS)3. Genistein and daidzein decrease monocyte chemoattractant protein-1 and collagen-induced platelet aggregation in a dose-dependent manner, indicating that they could act as antithrombotic and antiatherogenic agents [161].
Phytoestrogens also exert a direct effect on cardiac myocyte contractility, albeit in opposing ways; genistein exerts a positive inotropic effect on isolated ventricular guinea pig myocytes, whereas equol exerts a negative inotropic effect [162]. This suggests that both of these soy components might possess distinct mechanisms of action, and the cumulative effect of these phytoestrogens on cardiac function might be the result of specific dominating actions of either genistein or equol.
There is evidence in the literature that the hormonal effects of phytoestrogens can affect fertility in humans and animals of both sexes. Genistein causes increased cAMP stimulation, leading to acrosomal loss and an increased rate of capacitation in human sperm populations, resulting in an inability of the sperm to fertilize an ovum [163]. When genistein and daidzein are tested in combination, their adverse effects on human sperm are more pronounced [163]. Male rats have decreased sperm production [164] and female mice produce fewer offspring [165] when exposed to high levels of phytoestrogens that are similar to those found in soy-based rodent chow. In order to lower reproduction during drought, quails have evolved to eat high-phytoestrogen-containing plants [166]. In addition, cheetahs in captivity were infertile until soy was reduced in the diet [167]. Phytoestrogens may also exert other detrimental hormonal effects; genistein has been demonstrated to increase glucose sensitivity of pancreatic islets in a dose-dependent manner (10–100 μM), and this effect was independent of ER. Furthermore, genistein has also been indicated to cause goiter in humans and animals by inhibiting thyroid peroxidase, which is also independent of ER [168]. Hooper
In addition to being an ER agonist, genistein in the μM range is also a potent tyrosine kinase inhibitor (TKI), whereas daidzein is inactive in this respect. Recent data indicate that several small molecule TKIs (e.g., anthracyclines, cyclophosphamide and trastuzumab) used to treat various cancers, are associated with severe cardiotoxicity, which might affect survival more than the malignancy itself [170,171]. The exact mechanism by which these TKIs induce cardiotoxicity remains under investigation. The relevance of genistein as a TKI has been most extensively studied with respect to ischemia. In rat hearts, ischemic preconditioning, which offers cardioprotection, is completely abolished by the TK inhibitory action of genistein, by preventing the enhancement of phospholipase D activity [172]. In addition, preconditioning of the rat heart stimulates protein kinase C, MAPK, and MAPKAP kinase 2 activities, which are also inhibited by genistein [172]. A similar abolition of the protective antiarrhythmic effect of preconditioning was also observed in hearts perfused
There are conflicting data regarding the beneficial or adverse effects of phytoestrogens, depending on experimental conditions
The physiological effects of phytoestrogens are further confounded by the equol producing status of humans. There is some evidence in the literature suggesting that the equol-producing status of individuals might affect their response to dietary interventions using soy. A 2-year study on lipids in normo- and hypercholesterolemic postmenopausal women found that soy had the greatest hypocholes-terolemic effect in equol-producers compared with nonproducers [181]. Hence, it might prove informative to measure serum equol levels of bacteriotype individuals for their equol-producing ability before dietary interventions with phytoestrogens are undertaken.
Conclusion & future perspective
Sex by itself is an independent variable affecting every aspect of healthy and failing heart function. Why do women before the age of 65 years have a lower incidence of heart disease compared with men? There are compelling clinical and experimental data supporting the cardioprotective role of estrogen in conferring the female advantage in heart disease. However, the interaction between sex and CVD is affected by a plethora of extrinsic and intrinsic factors, and so cannot be attributed to a single factor such as estrogen. A clinical trial that was designed to test the benefit of treating men with a predisposition to heart attack with large doses of estrogen resulted in gynecomastia, impotence and venous thromboembolic disease [182]. Recently, men with higher levels of circulating estrogen were found to be associated with the highest level of cardiovascular risk, as they demonstrated high levels of LDL-cholesterol and low HDL-cholesterol [183]. Why natural, endogenous estrogens that are generally seen as cardioprotective in women increase cardio-vascular risk in men remains to be explained. The role testosterone plays in CVD, and how it interacts with sex, age, blood pressure and other potential confounding factors, is also unclear. Furthermore, a better understanding of the interaction of sex hormones with their cognate receptors, and their modulation during heart disease, will broaden our understanding of these hormones and the role they play in the cardiovascular sexual dimorphism.
A growing interest in alternative therapies to HRT for the treatment of HF and postmenopausal symptoms has led to a spike in the use of environmental agents that mimic endogenous hormones (i.e., phytoestrogens). Several confounding factors, such as lifestyle and demography, should be considered before we draw conclusions regarding the beneficial effects of phytoestrogens. Van der Schouw
Executive summary
After puberty, the left ventricular (LV) mass:body weight ratio is 25–38% greater in men compared with women. The increase in LV mass is primarily due to changes in myocyte size (hypertrophy) rather than their numbers.
The myocardial mass, myocyte number and average cell diameter and volume in women are preserved through the ages of 20–95 years. By contrast, men between the ages of 17–89 years show a gradual loss of nearly 1 g/year of the myocardium, accounting for approximately 64 million cells lost from both the left and right ventricles. In the remaining cells in males, myocyte cell volume increases at a rate of 158 μm3/year in the left and 167 μm3/year in the right ventricle.
There is no correlation between age and cardiomyocyte apoptosis between the two sexes. However, at any given age, cardiomyocyte apoptosis is threefold higher in men compared with women.
The force of contraction in a normal myocardium is comparable between males and females. However, the durations of contraction and relaxation are usually shorter, and the maximum velocities of tension development and decline are markedly faster in the female myocardium. The female myocardium seems to possess a lower myofilament Ca2+-sensitivity than the male myocardium.
Sex-based differences in the expression of myocardial Ca2+-handling proteins (the Na+-Ca2+ exchanger and sarcoplasmic reticulum calcium ATPase) have a direct impact on the functional changes in contractility.
Cardiovascular disease manifests itself differently in women compared with men. Myocardial infarction, hypertrophy, arrhythmias and fibrosis demonstrate sexual dimorphism in their manifestation and prognosis.
A sex-based dichotomy exists with respect to cardiac adaptation to exercise. Women tend to exhibit a relatively smaller increase in LV ejection fraction in response to exercise compared with men.
Women tend to increase their cardiac output primarily by increasing end-diastolic volume index without significantly increasing ejection fraction, compared with men who primarily decrease end-systolic volume index and raise ejection fraction.
Data from mice also suggest that exercise provokes a sex-dependent cardiac adaptation. Females tend to benefit more from exercise compared with males.
Potential candidates that mediate exercise-induced cardiac hypertrophy include CaMKII, Akt and GSK-3β.
The exercise-mediated beneficial effect of HSP 70 is influenced by estrogen.
Women have been under-represented in the majority of the cardiovascular clinical trials in the past, which has left many questions unanswered in interpreting the efficacy of various drugs in women.
A number of cardiovascular drugs, such as digoxin, statins, Ca2+-channel blockers and acetylcholineesterase inhibitors, exhibit differential effects in men and women in terms of efficacy, pharmacokinetics or adverse drug reactions. Therefore, it is very important to study cardiovascular drugs in both sexes.
Men and women both produce significant and physiologically relevant amounts of estrogen and testosterone.
Estrogen exerts cardioprotective effects via several mechanisms.
Testosterone has been demonstrated to have both protective and deleterious effects on heart.
Functional sex steroid hormone receptors; estrogen receptor (ER) α and ERβ and androgen receptors (ARs) are expressed in the heart and play important roles in cardiac pathophysiology.
ERα and ERβ play a protective role, especially in females, after various cardiac insults.
ERβ may be responsible for the antihypertrophic effects of estrogen.
Little is known regarding the androgen receptor-knockout or aromatase-knockout mice.
Estrogen is protective in volume and pressure overload in females. Testosterone is detrimental to females post-myocardial infarction.
HRT does not decrease heart disease risk, but does increase the risks of stroke, breast cancer and pulmonary embolism.
Phytoestrogens daidzein and genistein are naturally occurring isoflavones that are found in numerous edible plants, especially soybeans. Daidzein undergoes further metabolism by means of the gut microflora to obtain a biologically active metabolite, equol.
Phytoestrogens are structurally similar to estrogens and may act as both estrogen agonists and antagonists.
Phytoestrogens can both positively and negatively influence cardiac function.
Footnotes