
Abstract
Breast cancer remains a significant public health problem despite advances in the understanding of the molecular and cellular events that underlie the disease. Crucial pathways regulating the cell cycle, proliferation and survival of breast cancer cells have been investigated and aberrant components of these pathways have been exploited as new drug targets. However, the mortality from breast cancer is only slowly declining. Recently, a model has been proposed that might explain the heterogeneous biological features of breast cancer cell populations and their differential response to therapeutic agents, which has interesting implications for further progress in therapy. This model links the emergence of breast cancer cells to stem cells and progenitors, an observation originally made in other cancer entities. It hypothesizes that the tumors originate from a small population of undifferentiated cells. These cells can undergo self-renewal and are able to generate a large number of partially differentiated cells, which constitute the bulk of the tumor. These cancer stem cells resemble adult stem and progenitor cells found in the normal breast, but are deregulated in their patterns of proliferation and differentiation. They could originate from normal stem cells or from more differentiated progenitors and lose their normal growth restraints through a series of oncogenic mutations that deregulate a small number of central signaling pathways. If breast cancer really is a stem and progenitor cell disease, this will have important implications for the understanding of the emergence of cancer cells. A combination of the cell-type of origin, stem cells, early or late progenitors and the particular oncogenic mutations acquired could provide a new classification of the different types of breast cancer. These parameters might determine the mechanisms of cancer progression and the responsiveness of patients to drug treatment. Stem cell-specific features could possibly be exploited as innovative drug targets.
Keywords
Breast cancer is the most common cancer among women in Western Europe and North America, and its prevalence is increasing in developing countries [1]. A number of risk factors are known, among them age, genetic susceptibility, ethnicity, diet, socioeconomic status and reproductive history [2,3]. Among the dietary risk factors, only alcohol intake has a consistent and strong positive association; being overweight constitutes an additional risk [4]. A full-term pregnancy early in life is associated with a reduced risk for the development of breast cancer [5]. Owing to the multiple and diverse inputs contributing to breast cancer etiology, it is not unexpected that this is not a single disease. Histopathological analyses indicate a broad variety, and distinct subtypes are associated with different clinical outcomes [6–8]. The different responsiveness of the subtypes and the underlying molecular diversity are serious obstacles for the development of targeted cancer drugs. Tumor initiation and progression are most probably driven by acquired genetic alterations. These genetic and epigenetic changes have consequences for the autonomous growth behavior of the tumor cells by influencing the paracrine interactions with the cells of the tumor microenvironment and may also have endocrine effects [6,9].
A cellular hierarchy governs the relative distribution of cell types within the mammary gland [10,11]. A small population of cells with self-renewal properties maintains the tissue architecture and remodeling, and these fulfill the definition of stem cells [12]. The stem cell hypothesis has been extrapolated to tumor tissues and the postulation of the stem cell hypothesis has many attractive conceptual and practical implications. Cancer cells are distinguished by multiple mutations that alter their cell-cycle regulation, their sensitivity to apoptotic signals and their lifespan. Since stem cells persist within the organism for long periods of time, they have the potential to accumulate genetic damage and propagate this damage to their daughter stem cells and their downstream progenitors. Many tumors contain subpopulations of tumor-initiating cells that are able to reconstitute a tumor upon transplantation; these are called cancer stem cells (CSCs) [13]. CSCs share the characteristics of normal stem cells (i.e., they can self-renew and they can give rise to heterogeneous cell populations within a tumor and maintain the tumor mass) [14]. They are also more resistant to chemotherapeutic drugs and radiotherapy than their progenitors and thus represent a cellular reservoir for tumor recurrence [15,16]. CSCs can be derived directly from mutational events in stem cells that confers proliferative properties to the CSCs or they can result from mutations in progenitors that provide the CSCs with their self-renewal capacity. Both pathways can be used and result in tumors with different pathological properties [17,18]. The characterization of the signaling pathways that provide self-renewal properties to CSCs and the targeted interference with limiting signal-transduction components offer promising new therapeutic approaches for the treatment of cancer.
It is reasonable to assume that breast cancer might originate from a CSC population. This cell population is functionally similar to stem cells in the normal mammary gland [19,20]; however, the identification and characterization of the CSC population has not advanced far. Under particular experimental conditions, it is possible to functionally demonstrate a small population of cells with self-renewal capability. This has important biological and clinical consequences, and the interpretation of tumors as hierarchical cellular structures has profoundly influenced our picture of the emergence and progression of breast cancer.
Evidence for the presence of stem cells in mammary tissue
The perpetual renewal of tissues and organs is driven by resident stem cells and progenitors. These cells guarantee tissue maintenance and regeneration following injury or involution, and most tissues and organs contain small populations of primitive stem cells and progenitors. Both stem and progenitor cells have an important role in fetal development and contribute to the generation of tissues and organs. Stem cells are positioned on top of the cellular hierarchy and give rise to progenitors with more restricted lineage potential. The stem cells can divide and self-renew to generate daughter stem cells, or they can differentiate into a variety of mature cell types [21].
The application of the stem cell hypothesis to tumor tissues is persuasive and it explains important observations, particularly the heterogeneous cell types and morphologies of cells present in tumors, the different potential of tumor cell subfractions to re-establish tumors upon transplantation and also the differential sensitivity of cells within tumors to chemotherapy and radiation therapy [22,23]. In addition, the course of disease and the progression and metastatic potential of tumors could possibly be explained by the particular genetic alterations that drive stem cell self-renewal and differentiation [24]. Despite these important implications, the isolation and characterization of normal stem cells or CSCs in breast tissue remains preliminary. Most of the evidence is based on the functional properties of a small fraction of cells, rather than on histological or biochemical studies.
The cyclical nature of mammary gland growth and involution initially suggested the presence of stem cells in this organ [12]. The ducts of the epithelium are formed by a layer of luminal epithelial cells surrounded by basal myoepithelial cells and the basement membrane. The primitive branched ductal system present in virgin animals expands during pregnancy, stimulated by the systemic release of steroid and peptide hormones. Side-branches and alveoli are formed, and secretory luminal epithelial cells fill the whole fat pad at parturition. Milk is produced during the suckling period, and the termination of suckling is followed by involution of the gland through massive apoptotic cell death. A ductal structure resembling that found in the mature virgin gland is restored. Thus, each cycle of pregnancy is accompanied by proliferation, differentiation and involution, a process driven by resident stem or progenitor cells [25,26]. The number and the fraction of mammary stem cells (MaSCs) seems to increase during pregnancy and decline again during involution [27].
The functional description of the mammary gland has been complemented by a partial molecular characterization of the main cell types involved in its structural organization (Figure 1). The mammary epithelium consists of ducts and alveoli that are composed of basal and luminal cell layers. The basal cell layer comprises myoepithelial cells and harbors the mammary epithelial stem cell compartment. In mice, the luminal cell layer is composed of two functional lineages that are distinguished by the expression of the cell surface proteins CD24 and Sca-1 [28]. Lin−CD24+/highSca-1+ luminal cells express estrogen receptor α (ERα) and the prolactin (PrlR) and progesterone receptors. The Lin−CD24+/highSca-1− luminal cells lack expression of ERα. These cell types emerge from stem cells [29]. Some of the signals that cause the generation of myoepithelial, luminal ERα− and luminal ERα+ daughter cells and that control cellular homeostasis, fate determination and lineage commitment in the mammary gland have been identified [30]. Paracrine cellular interactions, transcriptional regulators and epigenomic modifications have been implicated in these processes [10].
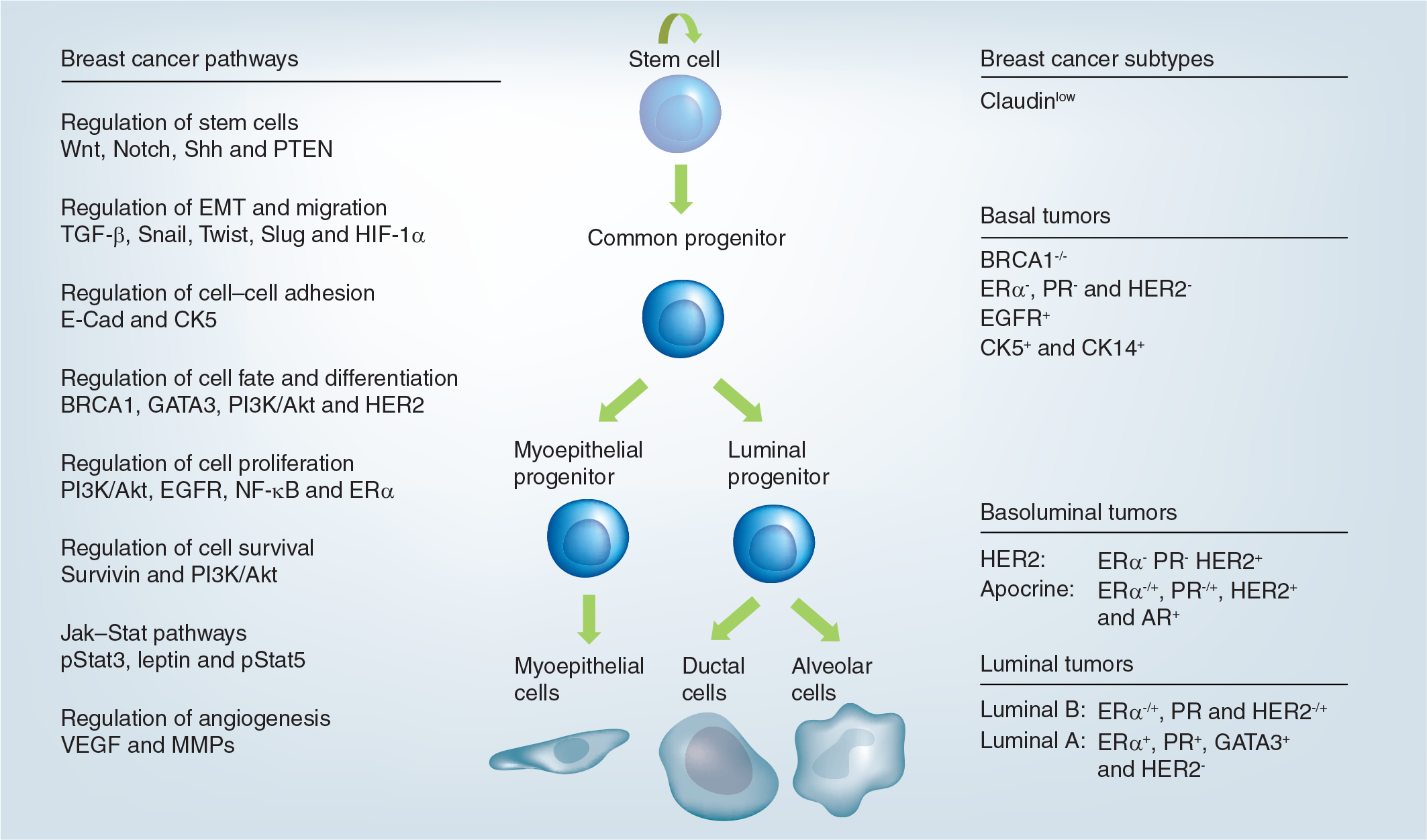
Epithelial cell hierarchy in the mammary gland, breast cancer subtypes and breast cancer pathways.
For example, the transcription factor GATA3 specifies commitment in the general luminal lineage [11] and Elf5 has the same effect on alveolar cell fate [31]. Wnt-4 is a signal protein that acts in a paracrine fashion downstream of the progesterone receptors and is involved in ductal side-branching [32]. Amphiregulin, produced by ERα+ cells in response to estrogen, stimulates mammary stem cell activity [33] and Notch signaling regulates luminal cell fate [34].
More direct evidence for the existence of adult stem cells in the mammary gland is based on transplantation experiments [35]. The emerging mammary tree can be efficiently removed in very young mice and these cleared fat pads can be inoculated with exogenous mammary cells. Small fragments of mouse mammary tissue and very small numbers of marker-enriched cells or unfractionated lentivirally transduced total mammary epithelial cells [Groner b et al., Unpublished Data] were able to reconstitute the epithelial component of the mammary gland and support its outgrowth and differentiation [36]. Although these stem cells have not been unequivocally identified, they have been found to be associated with particular cell-surface markers. Cells sorted for CD24 and CD29 (β1 integrin)/CD49f (α6 integrin) were found to be enriched in cells with a repopulation capacity, but they are not necessarily stem cell-specific [11]. Mouse CD24 is a marker expressed on epithelial cells and merely allows the distinction between epithelial and stromal components.
CD29 has been used to enrich stem cells from different tissues and could possibly establish a link between stem cells and their niche [37]. Integrin subunits form heterodimeric receptors that interact with glycoproteins present in the extracellular matrix or on the surface of neighboring cells. Integrins detect receptors in the environment of the cell and confer intracellular signals through kinases, scaffolding proteins or small GTPases. Basal cells and basal membrane components probably cooperate to establish the stem cell niche [38]. The targeted deletion of the CD29 gene in mammary basal cells results in the loss of the functional stem cell population and affects lobular-alveolar development [37].
Since the stem cells are mainly functionally defined through their ability to repopulate cleared fat pads, different cell populations can be investigated for the presence of stem cells. The highest proportion of stem cells has been found in the terminal end buds of the developing mammary gland in virgin mice, while only a few stem cells have been detected in the alveoli of lactating mice [12]. Cell-surface markers can be used to enrich stem cells by flow cytometry (e.g., fluorescence-activated cell sorting) and to remove hematopoietic and endothelial cells from the cell population. The combination of Lin−CD24medSca-1lowCD29high/CD49fhigh expression has led to the partial purification of cells with repopulating potential in mice; this subpopulation constitutes approximately 1–2% of all cells [36,39].
The fraction of cells within the entire mammary epithelial cell population that conforms to the definition of stem cells and is able to support repopulation of cleared fat pads has yet to be precisely determined [10], with estimates that range from approximately 0.02 to 1% [36,39,40]. The experimental procedures required to enrich the stem cell fraction involve tissue dissociation and flow cytometry, as well as the functional assays based on fat pad transplantation and the outgrowth of ductal trees. The different yields of repopulating cells reported might reflect the different procedures and reagents employed by individual groups [35].
These observations were initially made with mouse tissues but then were extended to investigate human MaSCs in breast tissue. The markers that have been employed in fluorescence-activated cell sorting experiments include the epithelial cell adhesion molecule, EpCAM, CD49f and the luminal cell-specific glycoprotein, MUC1 [26]. EpCAM and CD49f, are epithelial cell-specific, are expressed to different extents by luminal and basal cells and reconstituting cells have been found in the Lin−EpCAMlowCD49fhighMUC1− cell population. They were tested for their ability to engraft into the renal capsule. Alternatively, mammary cells of human origin can be transplanted into humanized mouse fat pads, which are fat pads conditioned with human fibroblasts [8]. The detoxifying enzyme aldehyde dehydrogenase (ALDH) 1 has also been identified as a marker for these stem cells [41]. Additional fractionation experiments were carried out with dispersed cells from human breast tissue. Bipotent progenitor cells (Lin−EpCAM+/CD49fhi/CALLA[CD10]+/Thy1+/CD133−), luminal committed progenitor cells (Lin−EpCAM+/CD49f+/MUC1+/CD133+/CD10−/Thy1−) and differentiated luminal cells were identified by surface markers from normal human breast tissue. ERαlow/PRhigh mRNA expression was found in the bipotent cell population and ERαhigh/PRlow mRNA expression was found in the luminal-committed progenitor population [42].
Breast cancer stem cells
Cancer cells are thought to originate through the accumulation of multiple mutations, a process that requires many cell divisions. The self-renewal capacity of stem cells is a prerequisite for the maintenance of progressive genetic changes and allows mutations to be propagated through cellular generations. The activities of tumor suppressor proteins might at least temporarily counterbalance detrimental mutations [43,44].
The functional properties of stem cells, which comprise self-renewal and the generation of proliferating progenitor cells and differentiated tissue cells, has been extended to the investigation of tumors [45]. In one study, tumor cells were dispersed and separated according to particular cell-surface markers and the resulting subpopulations were investigated for their potential to form tumors upon transplantation into immunodeficient recipient mice [46]. Only a subpopulation of cells obtained from human breast cancers is able to cause tumor growth when introduced into nonobese diabetes/severe combined immunodeficient mice. These cells are distinguished by the expression of Lin−CD44+CD24−/low and their fraction varies when individual tumors are compared. These cells represent between 1 and 20% of the total cancer cells [20,25]. Cells expressing Lin−EpCAM+CD44+CD24−/low were most effective in causing tumor growth upon transplantation into mice and demonstrated a phenotypic heterogeneity similar to that found in the parental tumors. Breast CSCs were also found in established cell lines. However, a correlation between the markers described previously was not necessarily found [15,47].
The subfractionation experiments combined with the transplantation of the sorted cells into immunocompromised mice indicate that the Lin−CD44+/CD24−/low cell population is enriched in tumor-initiating cells, but these markers do not provide for the purification of CSCs. Additional markers, such as ALDH1, have been included to further delimit the stem/progenitor cell population [41]. As with normal tissue stem cells, tumors might harbor specific cell populations that undergo self-renewal as well as various degrees of differentiation. CSCs could arise from normal stem cells through oncogenic transformation or from more developmentally advanced, replication-competent progenitor cells, possibly through the deregulation of self-renewal pathways. The cell population that results is characterized by distinct genetic and epigenetic changes that gives rise to the cellular heterogeneity of tumors. Instead of generating ductal and alveolar cells, the CSCs undergo aberrant and limited differentiation. Their origin and the peculiarities of the acquired mutations cause the different breast tumor subtypes and breast cancer heterogeneity [48]. Alterations in gene expression patterns can also be caused by epigenetic changes and cell–cell interactions that define the stem cell niche.
Genome-wide mutational analyses have yielded a large number of mutated genes implicated in human breast cancer cells [49]. Despite the large number of mutated genes, many of the consistently altered genes can be assigned to particular molecular pathways affecting cell adhesion, intracellular signaling, DNA topological changes and cell-cycle control [50]. The large number of mutations accompanying the progression of human tumors could be the reason for the substantial heterogeneity observed even within the individual tumor subclasses [51], and may reflect the differences in the clinical course of the disease in individual patients [52].
Mouse models have demonstrated the close relatedness of tissue stem cells and CSCs. Tumors induced by targeted oncogene expression in the mammary gland, mouse mammary tumor virus (MMTV)-Wnt and MMTV-polyoma middle T transgenic mice or p53 deletion, demonstrated that CSCs and MaSCs share similarities [53–55]. The mammary luminal progenitor marker CD61 identifies CSCs in the MMTV-Wnt-1 mouse model of mammary tumors [56]. Human MaSCs are sensitive to oncogenic mutations. HER2 expression, activation of Notch and Hedgehog signaling and loss of expression of BRCA1, enhance their engraftment in immunodeficient mice [57]. Human repopulating cells or progenitor cells can function as a source of CSCs [58].
Although CSC-transplantation experiments have generated much enthusiasm, gaps in the interpretation of these data still remain. When established human breast cancer cell lines were sorted according to their CD44, CD24 and epithelial-specific antigen (ESA) expression, the fraction of Lin−CD44+/CD24−/low cells did not correlate with their tumorigenicity. However, a small number of isolated Lin−CD44+/CD24−/low/ESA+ cells were able to form tumors upon transplantation into mice [15]. In this study, it was found that luminal breast cancer cell lines (MCF7 and SUM225) are nearly 100% ESA+ and that, in these cell lines, the selection of Lin−CD44+/CD24− cells coincides with the enrichment of tumorigenic cells. In basal cell lines (MDA.MB.23I, SUMI59 and SUM1315), a high percentage exhibit a Lin−CD44+/CD24− phenotype, and the selection of ESA+ cells is further enriched for tumor-initiating cells. In cell lines with two distinct CD44/CD24 populations (SUM149), the Lin−CD44+/CD24−/ESA+ subpopulation contains the tumor-initiating cells. These results are in accordance with clinical observations indicating that just the relative abundance of CD44+/CD24−/low cannot be linked to tumor progression or event-free and overall survival [59]. Transplantation experiments with single cells from human lung and breast cancer cell lines demonstrated that the majority of the cells had tumor-initiating potential, indicating that the relative proportions of tumor-initiating cells in these lines can exceed the fraction of stem cells found in normal tissues by far [60].
Signaling pathways responsible for stem cell properties of cancer stem cells & their implications for therapeutic interventions
Cellular phenotypes are usually determined by extracellular cues that act through the mediation of cell-surface receptor systems and the induction of intracellular signal transduction pathways. Therefore, it is not unreasonable to assume that CSC properties are induced and maintained by pathways that are able to self-renew, and the generation of partially differentiated progenitor cells [61]. The involvement of these pathways in human breast cancer has not yet been investigated in sufficient detail. However, the functions of several distinct signaling pathways in stem cell biology are well established. Most of these pathways have initially been discovered by genetic analysis in Drosophila and have subsequently been investigated in mammals. Their deregulation in mice has been demonstrated to cause mammary tumors, and their activation has been discovered in human tumors [19,21]. Among them are pathways triggered by the aberrant overexpression of HER2, the loss of PTEN or the activation of the Wnt, Notch or Hedgehog signaling.
The CSC hypothesis postulates that, upon asymmetric cell division, a cell with stem cell properties is retained and a second cell partially differentiates and gradually loses its repopulation potential. These processes must be controlled by signaling pathways that govern stem cell self-renewal and differentiation. A number of candidate genes and pathways have been taken into consideration. The HER2 receptor is a member of the EGF receptor family and is very strongly expressed in approximately 25% of human breast cancers. HER2-overexpressing tumors grow aggressively and are frequently associated with metastasis formation and an unfavorable prognosis [62]. HER2 overexpression is significantly correlated with the expression of the stem cell-marker ALDH1 and increases the proportion of stem cells [41,63]. Treatment of responsive tumors with the monoclonal antibody Herceptin® (trastuzumab) reduces the stem cell population. HER2 may regulate the stem cell population in breast tumors by preventing the progression of stem cells into ERα+ progenitor cells [63]. This drug was most successful when it was applied in adjuvant treatment of early breast cancer patients [64].
Another pathway that is frequently deregulated in cancer cells originates with the activity of phosphoinositide-3 kinase and results in the induction of the Akt kinase and mTOR. PTEN, a phosphatidylinositol phosphatase, is an intermediate pathway component and an enzyme that is functionally impaired in approximately 40% of the breast cancer cases. A correlative observation has been made regarding the self-renewal of hematopoietic and neuronal stem cells: they are regulated by PTEN [21]. Therefore, it is reasonable to assume that inhibitors of Akt and mTOR might also be useful therapeutics in breast cancer treatment.
The Wnt ligands activate a signaling pathway and induce transcription factor activities that regulate cell fate decisions, cell proliferation, morphology, migration, apoptosis and differentiation [65]. Wnt signaling can influence mammary gland growth, differentiation and possibly involution, and exerts an effect on the self-renewal and differentiation of stem cells [66]. Transgenic mice expressing a MMTV-Wnt oncogene develop mammary tumors that strongly express stem cell-markers [19,56].
Notch signaling regulates cell fate determination, survival and proliferation, and is involved in normal mammary development and differentiation [34,67–69]. Transgenic mice expressing a constitutively active form of Notch4 fail to develop normal mammary glands, but do develop mammary tumors. Notch signaling promotes self-renewal and proliferation of early progenitor cells and restricts myoepithelial lineage-specific commitment and proliferation. It also promotes branching morphogenesis, but has little effect on terminally differentiated mammary epithelial cells. Notch signaling may prevent terminal differentiation and maintain mammary epithelial cells in a proliferative state [57,67,68]. Notch3 signaling is particularly important for the proliferation of HER2-negative breast cancer cells [70]. Finally, Hedgehog signaling regulates the self-renewal of both normal and malignant human MaSCs. The pathway is mediated through the action of the polycomb protein, BMI-1 [71].
The Stat family of transcription factors are downstream mediators of multiple cytokine- and growth factor-initiated signaling events [72]. Two members of this family, Stat3 and Stat5, regulate crucial steps in the development of the normal mammary gland. The deregulation of their activities contributes to breast cancer [73]. Although both genes can exert oncogenic activities, Stat3 and Stat5 interact and influence each others' potential for target-gene transcription. Primary human breast tumors displaying activation of both factors are more differentiated than those with Stat3 activation alone and demonstrate decreased proliferation and increased sensitivity to the chemotherapeutic drugs [74]. IL-6 induces malignant features in Notch3-expressing stem or progenitor cells from a human ductal breast carcinoma line [75].
Targeted tumor therapy relies on genetic and biochemical distinctions between normal and tumor cells. The identification of specific signaling pathways and signaling components active in CSCs opens new possibilities for therapeutic exploitation. CSCs could possibly be targeted and these agents might improve conventional treatment results. However, CSCs have properties that make them more resilient against chemotherapy [16] and ionizing radiation [76]. New therapeutic agents that target the genuine stem cell properties need to be designed.
Agents could be developed that induce the differentiation of cancer cells. This is a strategy that is being employed in the treatment of acute promyelocytic leukemia. All-trans retinoic acid promotes promyelocytic differentiation [77] and has also been shown to affect breast CSC differentiation [78]. Alternatively, the elimination of CSCs could be achieved by targeting crucial regulatory genes and signaling pathways. The pathways regulated by PTEN, Wnt, Hedgehog and Notch are involved in the self-renewal of both normal MaSCs and CSCs, and constitute promising candidates. The role of Notch might not be restricted to self-renewal of MaSCs since Notch might also regulate luminal cell fate commitment [34,42]. The Hedgehog pathway can be inhibited with cyclopamine and is essential for the maintenance of CSCs in myeloid leukemia [79]. Inhibition of the Notch pathway was found to prevent CSC self-renewal and inhibits tumor growth in leukemia [80], but could possibly be extended to breast cancer [81]. Many of the signaling components involved in these pathways are conventionally considered as ‘non-druggable’ (i.e., they do not have enzymatic activities or small molecular weight compound-binding pockets). New approaches based on the inhibition of protein–protein interactions or protein–DNA interactions must be considered in order to target pathway end points [72,82].
Cancer stem cells are probably maintained in a particular cellular environment and this niche might itself be a drug target [18]. This is particularly important for CSCs in the breast and those that are frequently resistant to endocrine therapy [22]. In normal breast tissue, the stem cells have a basal phenotype and do not express ERα [29]. If this is also the case in breast cancer, CSCs may be endocrine-resistant and treatment responses are attenuated by paracrine effects emanating from neighboring ERα+ tumor cells. Normal breast epithelial stem cells are also dependent on EGF-receptor activation and other growth factor receptors. Increased growth factor-receptor activation in endocrine-resistant breast cancers is accompanied by an increased proportion of stem-like cells. This might be a consequence of the endocrine therapy [22]. Epigenetic regulation of gene expression and the influences of the stromal microenvironment might also be responsible. Epigenetic reprogramming agents might be useful in downregulating growth factor-receptor expression and increasing the proportion of ERα-expressing cells [22]. Breast cancer cells of luminal, intermediate and basal phenotypes have an increased proportion of CSCs (i.e., Lin−CD44+/CD24low/ESA+ expressing cells) when compared with hormone-sensitive luminal cancers [15].
The stability & plasticity of cancer stem cells
If stem cells and CSCs are dependent on signal transduction pathways triggered by external cues, it is reasonable to assume that they are located in a specialized regulatory microenvironment. The cells constituting this environment probably contribute to the components that control the fate specification of stem and progenitor cells. The bone marrow, for example, can function as such a microenvironment and provides the proper architecture, which is composed of osteoblasts, osteoclasts, bone marrow endothelial cells, stromal cells, adipocytes and extracellular matrix proteins. The elements of the microenvironment regulate survival, growth and differentiation of diverse cell lineages through the provision of cytokines, chemokines, proteolytic enzymes and adhesion molecules. Since the environment can change and exert different influences on stem cells, it is reasonable to suggest that stem cells are not fixed entities, but that they can emerge and disappear as a function of changing conditions in their surroundings [24].
Questions must be asked regarding how stringent these requirements are and if persistent signaling is a prerequisite for the maintenance of stem cell properties. Transplantation experiments of mammary epithelial cells into cleared fat pads yield interesting insights. Stem cells with repopulation capacity are present in any portion of the murine mammary gland throughout the lifetime of the animal [83]. Even the disruption of a postulated stem cell niche, the dispersion into single cells and the infection with retroviruses does not abolish the stem cell potential upon transplantation [34,84]. Mixing experiments, in which distinct mammary epithelial progenitors were transplanted, suggested that the niche conditions met in the fat pad can trigger a redirection of the cell fate. The interaction with the mammary microenvironment in vivo could reprogram committed cells of even non-mammary origin to give rise to differentiated luminal and myoepithelial cells [85].
Epidemiological observations, linking pregnancy and the incidence of breast cancer, have led to investigations in which the influence of the mammary microenvironment on stem cell properties have been studied [86]. Cycles of pregnancy and lactation are associated with strong proliferation, differentiation and apoptosis of epithelial cells, and are regulated by the systemic levels of circulating hormones. Hormonal conditions also influence the relative risk of breast cancer formation. Early full-term pregnancy reduces the risk of breast cancer. Rodent models have been established in which the cooperation of estrogen and progesterone and carcinogen induction of mammary tumor formation has been demonstrated. Cellular proliferation is blocked as a consequence of hormone treatment, and the tumor suppressor gene p53 appears to play a crucial role in hormone-induced protection against tumor formation [87]. However, the effects of estrogen and progesterone on tumor formation are dependent upon the conditions of exposure. Extended periods of estrogen and progesterone exposure increase the risk of breast cancer, while short durations and doses, such as those encountered during pregnancies, reduce the risk of breast cancer. It is thought that the hormones cause changes in gene expression in the mammary epithelial cells that persist even after hormone withdrawal and induce a switch in the developmental fate of mammary cells [88].
Analysis of the cellular subtype composition revealed that so-called parity-induced mammary epithelial cells (PI-MECs), are only present in the lobuloalveolar units of animals after postlactational remodeling [89]. PI-MECs probably originate from the lobule-limited progenitor population with alveolar differentiation features, but contribute to ductal elongation throughout the ductal outgrowth upon transplantation into virgin hosts. They can self-renew and differentiate into epithelial populations expressing luminal and basal cell-markers, and form secretory alveoli. Thus, the PI-MEC cells assume the properties of multipotent stem cells. Changes in the hormonal milieu appear capable of influencing the number of cells with stem cell potential and could thereby exert an effect on tumor incidence [90,91].
Important observations have been made that suggest that transitions between cell types can occur and that these transitions have a role in tumor progression. Epithelial and mesenchymal states are not absolutely stable and transitions from one to the other contribute to tumor progression and intratumoral heterogeneity. Epithelial cells can reactivate a latent geneexpression program and undergo an epithelial to mesenchymal transition (EMT). EMT is triggered by growth factor signaling, tumor stromal cell interactions and hypoxia, and is mediated by transcription factors. The cells express stem cell-markers, transcription factors such as Snail and Slug and matrix metalloprotease 2, and exhibit the activation of the TGF-β signaling pathway [92,93]. The cells also lose luminal cell-markers and acquire basal–mesenchymal properties. The epithelial cells lose cell contacts and polarity, undergo cyto-skeletal changes and acquire increased motility and invasiveness. The EMT does not appear to be complete and is reminiscent of a luminal to basal transition. The cells still express CD24, a marker mainly expressed by epithelial cells and not by stromal cells.
Transformed epithelial cells with acquired mesenchymal traits are found in aggressive breast cancer subtypes. The EMT program also seems to confer stem cell properties on transformed cells and increases the proportion of cells that are able to initiate metastasis [94,95]. EMT promotes the development of therapy-resistant breast tumors [96]. ERα suppresses the expression of transcription factors regulating EMT [97].
We correlated the epithelial cell hierarchy in the mammary gland with breast cancer subtypes and the molecular pathways deregulated in breast cancer cells (Figure 1). The cell types found in mammary tissue, stem cells, progenitor cells and differentiated cells are hierarchically ordered. MaSCs give rise to common progenitors that further differentiate into myoepithelial- and luminal-restricted progenitors. These progenitors in turn produce luminal or basal myoepithelial cells. The alveolar luminal cells terminally differentiate into milk-secreting cells. A ductal system containing multipotent, committed ductal and alveolar precursor cells persists after involution. They develop into a fully functional epithelium in subsequent pregnancies. CSCs potentially arise from MaSCs through oncogenic transformation or from more developmentally advanced, replication-competent progenitor cells. The origin of the initially transformed cell, the peculiarities of the acquired mutations and the particular deregulated pathway act in concert and cause different breast tumor subtypes, basal tumors and basoluminal tumors. Additional breast cancer heterogeneity is found within these subtypes [49–52].
The clinical significance of cancer stem cells
The suspected roles of CSCs in the maintenance of tumors, their metastatic potential and their drug insensitivity suggest numerous therapeutic implications. It would be most advantageous if these parameters could be exogenously manipulated [98]. Targeted approaches to CSC functions should spare normal stem cells to avoid collateral damage. However, CSCs were initially found to be more resistant to conventionally applied drugs [99]. The increased resistance to drug treatment suggests that CSCs may be quiescent and thus escape many of the chemotherapeutic agents dependent on DNA synthesis. The expression of ATP-binding cassette transporters in CSCs might further enhance this resistance phenotype [100].
The discovery of drugs that specifically interfere with the unique functional properties of CSCs is made difficult by the low fraction of these cells within tumors and the absence of specific cell-surface markers that would enable their isolation. The induction of EMT in normal or neoplastic mammary epithelial cell populations results in the enrichment of cells with stem-like properties [95] and with increased resistance to chemotherapy drug treatment. This observation was integrated into an assay system to identify agents with specific toxicity for epithelial breast CSCs [101]. Compounds with selective toxicity for breast CSCs were found that reduce the proportion of CSCs, inhibit mammary tumor growth in vivo and induce increased epithelial differentiation of tumor cells.
The mechanism of action of the highly beneficial drug, Herceptin, a monoclonal antibody directed against the HER2 receptor and used in the treatment of breast cancer, has been linked to effects on CSCs. This drug was initially used for the treatment of metastatic breast cancer, but later was found to have remarkable effects on recurrence rates when used in the adjuvant treatment of HER2-positive early breast cancer [64]. It seems that HER2 exerts its unfavorable transformation phenotypes through its effects on CSCs. Its overexpression increases the proportion of stem cells, an effect that can be reversed by treatment with Herceptin [63].
The CD44 molecule is a marker observed on the surface of many stem cells. The interaction of CD44 with a specific monoclonal antibody has been exploited to eradicate acute myeloid leukemic stem cells [102]. This target molecule may also become valuable for the treatment of breast cancer. Genetic programs and signaling pathways differentially utilized in normal stem cells and CSCs might provide the best options. The targeting of Wnt signalling in skin tumors [103] and BMP expression in glioblastomas, causing the depletion of the CD133+ cell fraction and the induction of a more differentiated phenotype, might be beneficial.
Additional drug targets might be deduced from the sequencing of mRNA found in breast tumor cells and the identification of consistently mutated genes that cause human cancers. Technical advances in sequencing have made it possible to examine the genomes of cancer cells comprehensively and compare them with those found in normal cells. In addition, a relatively large number of mutated genes have been found in human cancers. Many mutations occur during tumor progression; individual tumor entities, such as colon and breast cancer, differ in the mutations found, and vast differences are even found in individual tumors of the same entity [50]. Although individual tumors differ strongly from each other, ‘driver mutations’ have been identified that can be assigned to a limited number of central molecular pathways [52]. Mutations in genes (e.g., those affecting the activity of the phosphatidylinositol 3-kinase pathway) were found in more than 30% of colon and breast cancers. This pathway is important for both cell growth and invasion. Other components affect the function of the TP53 (p53) and NF-kB pathways or inappropriately activate kinases, such as Akt and mTOR [104]. These sequencing studies are still not comprehensive and do not reveal alterations due to copy number changes, translocations and epigenetic modifications. They also do not take into account the cellular composition of tumor tissues. An extensive enrichment or, preferably, the purification of CSCs will be required in order to identify mutations by sequencing analysis that determine their phenotype. The heterogeneity found in tumors may reflect different properties specific to individual patients, and it will be interesting to find possible correlations with patient prognosis, response to therapy and overall survival. They will also spur the development of targeted therapeutics and enhance predictive potentials.
The frequent involvement of the tumor suppressor protein p53 in most human tumors has led to extensive studies concerning its functions. p53 is a stress-response protein, which suppresses tumor formation by triggering apoptosis or preventing damaged cells from proliferating, but recently p53 has also been linked to stem cell properties [105]. Disruption of the p53 network enhances the production of induced pluripotent stem (iPS) cells [106] and reduces the requirements for reprogramming to the expression of only two factors, Oct4 and Sox2. iPS cells have the same capabilities as embryonic stem cells, can self-renew and give rise to all of the tissue types of the body. The effect of p53 is mediated by the cell-cycle inhibitor, p21. The extent of the reprogramming process is remarkable. Viable mice can be derived from iPS cells obtained through tetraploid complementation from mouse embryo fibroblasts [107]. The pluripotent potential of these cells can be re-established, a property intimately connected with open chromatin. Highly accessible chromatin is essential for the unique properties of stem cells. Embryonic stem (ES) cells maintain an open chromatin structure and express a large proportion of their genes. Differentiation of ES cells into mature cell types is accompanied by heterochromatin formation and gene silencing. These characteristics are relayed by chromatin-remodeling proteins (e.g., Chdl). This protein is an essential regulator of open chromatin in stem cells and is essential for the maintenance of ES-cell pluripotency [108]. Reprogramming of adult tissue cells into iPS cells is similar to transformation. Cooperating genes generate a less differentiated cell with self-renewal capacities. It is conceivable that the loss of p53 could contribute to the acquisition of CSC-like properties of differentiated cells. This would increase the cell types that have a function in the origin of cancer formation and not limit them to only normal organ stem cells. The identification of the cooperating factors that maintain the differentiation potential of normal MaSCs or the tumor-forming potential of breast CSCs would be of great interest. A role of p53 in the regulation of the polarity of stem cell division has been described in a mouse mammary tumor model [55]. It appears that self-renewing, symmetric divisions of stem cells can frequently be observed in stem cells without p53 functions. This leads to increased numbers of stem cells in p53-deficient mice and favors the formation of mammary tumors.
Although the CSC niche is conceptually attractive as a target of interference and might offer novel possibilities to disrupt the cellular communication required for CSC maintenance, the components involved remain poorly defined. It is possible that remodeling processes in the tumor tissue alter the stem cell niche and promote the expansion of the stem cell pool, reminiscent of the developmental steps in the normal mammary gland under the control of systemic hormones [109]. The cell populations expand and regress drastically during puberty, pregnancy, lactation and involution, and the MaSCs and progenitors have to adapt to multiple local and systemic cues. Steroid hormones act in a paracrine fashion in the mammary gland, cooperating with locally produced factors to affect cell–cell interactions. The stem cell niches might change with the developmental stage and the hormonal milieu [38]. The CSC niches themselves might have aberrant properties owing to their adaptation to the requirements of CSCs. The simultaneous interference with the autonomously active tumor cell pathways driving their growth and survival, and interference of the supportive microenvironment, appears to be a promising strategy. Chemotherapeutic and antiangiogenic agents might function together effectively [110].
Treatment of tumors with chemotherapeutic and cytotoxic drugs or hormone antagonists puts selective pressure on the affected cells and triggers an evolutionary process. The tumor cells probably evolve in conjunction with the interacting cells in their microenvironment and might yield the outgrowth of adapted variants. The emergence of drug- and hormone-resistant tumors is a well-known consequence in many patients and might be linked to evolved CSC phenotypes. Tamoxifen-resistant ERα+ breast cancers exhibit a more basal phenotype with a reduction in E-cadherin expression [111]. They also have an enhanced motility, with upregulation of Src kinase, NF-kB activation and CD44 expression [112]. CSCs in colorectal cancers are substantially enriched following chemotherapy [113]. Drug resistance is often acquired through the enhanced expression of ATP-binding cassette transporters 2 and 5, and multidrug resistance protein 1. These proteins are also strongly expressed in CSCs and contribute to their chemotherapy resistance [99].
Future perspective
The presence and molecular characteristics of CSCs have interesting implications for the understanding of tumor etiology, their response to treatment and the development of novel therapeutics [92]. If CSCs are more resistant to conventional chemotherapeutic drugs and radiation compared with the majority of the cells present in solid tumors, and if they are the source of residual cells that cause recurrence of tumor growth after therapy, strategies and targeted drugs must be designed that are preferentially aimed at communication of CSCs with their microenvironment. The targets of such drugs could be components of the pathways that maintain breast CSCs, such as the HER2 [57], Notch [57], Hedgehog [114] and Wnt [115] pathways. Since many of these components are not susceptible to conventional drug action, new classes of drugs, able to disrupt distinct protein–protein or protein–DNA interactions, must be discovered and developed [72].
Executive summary
The cyclical nature of mammary gland remodeling during each round of pregnancy, lactation and involution suggests the presence of stem cells.
The capability of a selected subset of cells to establish a functional epithelium after cell transplantation into mammary fat pads provides experimental evidence for the existence of mammary stem cells (MaSCs). Murine MaSCs are currently enriched by FACS using Lin−CD24medSca-1lowCD29high/CD49fhigh, and human MaSCs are enriched by Lin−EpCAMlowCD49fhighMUC1−.
MaSCs persist through cycles of proliferation and cell death and maintain the mammary tissue architecture.
Cancer stem cells (CSCs) represent a subpopulation of cells in a tumor tissue. They can be enriched by surface marker selection and Lin−EpCAM+CD44+CD24−/low from human breast tumors, and effectively reconstitute tumors after transplantation into mice.
CSCs share characteristics with normal tissue stem cells. They are thought to be relatively quiescent, have self-renewal capacity and give rise to progenitor cells with a high proliferative potential but a limited lifespan.
CSCs are thought to arise from tissue stem cells through mutagenic events that provide them with enhanced proliferation potential, or from progenitor cells through mutations that abolish their restrictions on self-renewal.
CSCs are more resistant to chemotherapy and radiation than their progenitors and might therefore contribute to the persistence and recurrence of tumors following primary treatment.
Their low rates of proliferation and the activation of signaling pathways (e.g., those originating from the EGF receptor family, the stem cell-factor receptor, the Hedgehog, Notch and/or Wnt/β-catenin pathways) may function cooperatively.
The induction of antiapoptotic proteins, increased DNA repair capacities and ATP-binding cassette transporter-mediated drug efflux protect the CSCs from cytotoxic drugs and the effects of radiation. Since cytotoxic drugs exert a strong selective pressure on tumor cells, they cause the emergence of resistant cells adapted to their microenvironment. These aspects of CSCs can be targeted by new drugs and provide new combinatorial treatment possibilities.
The CSC hypothesis has stimulated the discussion regarding the cellular origins of cancer and has offered possible explanations for the variability in the responsiveness of individual patients to particular drug treatments, as well as for seemingly contradictory observations concerning treatment response and overall survival of tumor patients. The hypothesis has also linked aspects of developmental biology and tissue homeostasis with tumor biology, especially the connection between the ability of stem cells to self-renew and the ability of tumor cells to proliferate in an unlimited fashion that has indicated that similar molecular mechanisms might be active.
The Wnt, Notch and Hedgehog pathways were initially discovered in developmental studies in Drosophila, but were later found to be causally involved in tumor formation and to be able to allow the maintenance of CSCs. These pathways provide particular molecular targets that are inhibitable and thus become valuable for the development of new therapeutics.
The use of Herceptin® as an agent able to reduce the fraction of CSCs in combination with chemotherapeutic drugs in the adjuvant treatment of early breast cancer is an encouraging example. Other signaling pathways that might exert selective effects on CSCs are PTEN/Akt and NF-KB.
Akt and mTOR can be inhibited by specific kinase inhibitors, and several NF-KB pathway inhibitors are in development. These new agents, alone or in combination with other drugs, might affect CSC properties and improve the response of tumor patients to existing therapeutic regimes.
Despite these conceptual and practical advances, fundamental questions remain. The characteristics and the proportions of CSCs vary when individual tumors are compared. This might reflect the relative flexibility of cells to assume stem cell properties under the influence of particular microenvironmental conditions and the specific oncogenic alterations found in individual tumors. The adaptation of CSCs to selective pressures exerted by therapeutic agents might also contribute to CSC heterogeneity.
Consistent characteristics must be found and exploited to make CSCs clinically treatable.
Footnotes
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.