
Abstract
Endometriosis is an estrogen-dependent disease and estrogen-related pathways are imbalanced in women with endometriosis. One of the key enzymes in estrogen synthesis is aromatase. Inhibiting this pathway at several points is a promising idea for the treatment of endometriosis. The third generation of aromatase inhibitors is becoming more potent in efficacy, with fewer side effects than previous generations, but cotreatment with other hormones is needed to inhibit ovarian stimulation. Other components that promote estrogen synthesis such as COX-2 can also be potentially targeted. Selective estrogen-receptor modulators could also be interesting in view of their tissue-specific effect. However, all these new drugs are still in an early phase of development. At present, it is too early to conclude that aromatase inhibitors, COX-2 inhibitors or selective estrogen-receptor modulators really present any added value compared with the existing drugs that can be used to achieve hormonal suppression in the medical treatment of endometriosis.
Endometrium is a tissue with continuous regeneration according to the menstrual cycle. The function of this tissue is to provide the appropriate environment for the implanting blastocyst after decidual transformation. In this regard, there is cell–cell communication, rapid proliferation, invasion and response to hormonal regulation in the endometrium. Estrogen is a key component of these very complex and diverging processes. Estrogens initiate the growth and structural development of functional endometrial tissue, including formation of the stromal and glandular cell network, and angiogenesis. Estrogens activate genes with an estrogen response element (ERE) in the regulatory site of their promoter. In this review, we analyze new treatment strategies for endometriosis utilizing the estrogen-pathway inhibition concept at different intervention points.
Estrogen pathway
Estrogen production in normal pathways
The main sources for estrogen are ovarian granulosa cells in the ovarian follicles from women during their reproductive age. Around menopause, the adipose tissue partly takes over the estrogen production from the ovary. The individual estrogen production of a single adipose cell is small, but the accumulated amount is stored in the tissue to maintain a permanent estrogen level in the peripheral circulation.
The biochemical and genetic background of estrogen synthesis in association with endometriosis has been reviewed recently [1,2] and is shown in Figure 1. Understanding regular estrogen production is important to distinguish it from its pathogenic form in endometriosis and to recognize possible intervention points. Briefly, in the ovary, the first step of estrogen synthesis is the entry of cholesterol from the cytosol into the mitochondrion, regulated by the steroidogenic acute regulatory protein (StAR) [3]. In the mitochondrion, cholesterol is converted to androstenedione, which is catalyzed either to progesterone or to the biologically active estrogen, estradiol, by six enzymes. In a key step, androstenedione is converted to estrone. The catalytic enzyme for this step is aromatase (P450arom/gene CYP19A1), a member of the cytochrome family, located in the smooth endoplasmic reticulum. Estrone itself has only a low estrogenic capacity. It is then further converted by a hydroxysteroid dehydrogenase enzyme (17β-HSD type 1) to physiologically potent estradiol. Subsequently, estrogen is secreted into the peripheral circulation and is ready to act on its target tissues, such as the endometrial tissue. Inside the cell, estrogen binds to its receptor in the cytoplasm and is transported into the nucleus to activate endometrium-related genes. This activation results in estrogen-dependent cell growth. Estrogen excess is eliminated by 17β-HSD type 2, an enzyme that neutralizes estrogen by converting it back into estrone.

Regulation of the estrogen pathway in the ovary and in endometriotic lesions.
While the ovaries produce estrogen directly from cholesterol, adipose tissue produces – by the use of aromatase – estrone from the circulating androstenedione, which is then further converted to estrogen in other extraovarian tissues.
The key enzyme of the estrogen-synthesis pathway after cholesterol has entered the mitochondrion is aromatase cytochrome P450 (CYP). The presence of CYP mRNA or protein is a good indicator of estrogen production. The genetic background and regulation of the aromatase gene (CYP19A1) is well known [2,4,5]. The promoter region of the aromatase gene is reasonably complex. Several tissue-specific promoters (n = 10) regulate the expression of this gene, resulting in different hnRNA, which, with alternative splicing, then matures into the same mRNA form. The main consequence of distinct tissue-specific promoters is the different strength in transcription activation and different activation signals to which they can respond. In the ovary, promoter II acts as a regulator sequence that is inducible by prostaglandin E2 (PGE-2). The activation takes place via cAMP, activating the steroidogenic factor 1 (SF1), which, in cooperation with other coactivators (e.g., CCAAT-enhancer-binding protein [C/EBP]α and cAMP-response-element-binding protein [CREB]), induces aromatase expression. By contrast, in adipose tissue, promoter I.4 is in charge for the aromatase regulation. The promoter I.4 is coregulated by glucocorticoids and cytokines from the class I cytokine family (e.g., IL-6, IL-11, leukemia inhibitory factor, oncostatin-M and TNF-α).
Normal endometrium & estrogen
During estrogen stimulation, the endometrium undergoes proliferation. Endometrium contains estrogen receptors, the catalytic enzyme to reduce estrone to estrogen (17β-HSD-1) and another hydroxysteroid dehydrogenase enzyme (17β-HSD-2) that controls estrogen levels by converting it back to estrone. Other gene products with a key function in estrogen synthesis, such as aromatase, are not traceable or are only traceable at a very low level (StAR) in the endometrium from women without endometriosis (Table 1) [2,6–8]. The other, nonregulational enzymes of the estrogen pathway seem to be present in a housekeeping manner [8]. The high level of regulatory proteins, such as chicken ovalbumin upstream promoter transcription factor (COUP-TF), Wilms tumor-1 (WT-1) and C/EBPβ, keep the estrogen synthesis shut down (Figure 1).
The expression of aromatase and other key enzymes related to estrogen synthesis in endometriosis tissue and in endometrium from women with and without endometriosis.

RNA-based method.
lmmunochemical-based method.
CYP: Cytochrome P450; HSD: Hydroxysteroid dehydrogenase; StAR: Steroidogenic acute regulatory protein.
At the end of the menstrual cycle, the endometrium begins to become necrotic and the menstrual endometrial tissue is cleared from the body through the vagina, but can also regurgitate into the peritoneal cavity in 70–90% of cases [9]. In healthy women, this regurgitated endometrium is thought to be cleared up by immune scavenger mechanisms, but this has never been substantiated by scientific evidence.
Estrogen & its relationship with endometriosis
Endometriosis is defined as the growth of endometrium outside the uterine cavity or myometrium. This endometriotic tissue is characterized by local estradiol biosynthesis, in contrast with the eutopic endometrium derived from patients without endometriosis.
Estrogen production in endometriotic lesions versus eutopic endometrium from control patients
As discussed earlier, in order to produce estrogen, eutopic endometrium from disease-free women only expresses enzyme (17β-HSD-1) to convert estrone to estradiol estrogen [2,6,7] and express some cascadial enzymes in a housekeeping manner [8]. By contrast, endometriotic lesions are equipped with all the enzymes needed for estrogen synthesis from cholesterol to estrone and the final step to estradiol conversion [2,8,10]. In endometriotic lesions, estrogen is synthesized in stromal cells induced by PGE-2 via the cAMP pathway, such as in the ovary [6,11].
In vitro, PGE-2 or cAMP at a low level are capable of turning on CYP aromatase expression in endometriosis-derived stromal cells, but not in cultured endometrial cells from disease-free women [6,12]. Indeed, aromatase is expressed in endometriotic lesions in vivo, but is undetectable in normal endometrium, peritoneum or decidua (Table 1) [7,10,11,13–16]. It is also worth noting that aromatase expression appears to be cycle dependent (more pronounced during the secretory phase) in some patients, but not in others [16]. In some endometriotic lesions, up to 39% in one study [16], aromatase expression is undetectable when assessed by immunohistochemical staining and aromatase-enzyme activity. These characteristics might indicate that these aromatase-lacking endometriotic lesions have a different (possibly less active) biological activity.
By analyzing the activating mechanisms that regulate gene transcription, it turns out that at promoter II, several differences can be observed between eutopic endometrium from women without endometriosis and those with endometriotic lesions [15,17–19]. In the eutopic endometrium of disease-free women, COUP-TF inhibits the expression of P450arom, SF-1 transcription factor is not present (even in stimulated conditions), and other repressors are also present. However, in endometriotic tissue, SF-1 is expressed and over-rules the COUP-TF suppression, which is downregulated in a competitive way [20]. The SF-1 repressor WT-1 also shows a lower level in endometriotic tissue compared with normal endometrium from nondiseased women [15,21]. The main regulatory problem may be even more upstream on the signaling pathway, towards the COUP-TF WT-1 direction and the genes involved in the regulation of estrogen synthesis [14]. Indeed, PGE-2 affects not only the expression of aromatase, but the whole estrogen pathway. Activated genes in endometriotic lesions range from StAR [22], which regulates the cholesterol entry to the mitochondrion, to genes involved in the catalytic pathway. Other coactivators are also elevated, such as C/EBPα, which cooperates with SF-1 to induce aromatase. However, C/EBPβ, known to repress the expression of SF-1, is scarcely present in endometriotic lesions [14].
Not only are estrogen-pathway gene-repression mechanisms abnormal in endometriotic tissue, but endometriotic lesions also lack the 17β-HSD-2-catalyzed reaction that converts estradiol back to its inactive form (estrone) [23]. Furthermore, endometriotic stromal cells are resistant to progesterone because of the under-expression of progesterone receptor-β [15,24,25], and therefore are not sensitive to the PGE-2-stimulated inhibitory effect of progesterone on estrogen synthesis that is observed in the endometrium of women without endometriosis.
Other mechanisms may also support the development of endometriosis. Indeed, via a positive-feedback effect, estrogen can upregulate PGE-2 via the estrogen receptor [26–28]. PGE-2 can in turn increase estrogen production from a low initial level to a higher level, allowing ectopic endometrial tissue survival, and perhaps the formation of endometriotic lesions. PGE-2 has a positive feedback on its own production by upregulating COX-2 in prostaglandin synthesis. Cytokine IL-1β also affects PGE-2 levels by stabilizing the mRNA structure of the COX-2 gene in endometrial stromal cells. Furthermore, IL-1β not only stabilizes, but also increases COX-2 expression in endometriosis [29,30]. Other cytokines such as IL-6, IL-11 and TNF-α also stimulate the cAMP level and might thereby stimulate aromatase activity in endometriotic tissue [31–34]. Based on the currently available evidence, it appears that endometriotic lesions are characterized by both an activation of the complete estrogen-synthesis pathway, and an absence of proteins that are able to inactivate estrogen locally.
Some alterations have been discovered in protein expression regarding the estrogen-synthesis pathway in eutopic endometrium from women with endometriosis compared with nondiseased controls. Whereas aromatase expression has been reported to be absent in eutopic endometrium from nondiseased women, expression of aromatase is detectable in eutopic endometrium from women with endometriosis, but at much lower levels than in the endometriotic lesions (see Table 1 for references). Indeed, COX-2, a key enzyme in prostaglandin synthesis, shows elevated levels in eutopic and ectopic endometrium from patients with endometriosis compared with endometrium from controls [29,35–39]. However, PGE-2 itself is produced in a cyclic manner during the cycle and is not abnormally expressed in endometrium from women with endometriosis compared with controls [40].
Recently, a working hypothesis based on the retrograde menstruation theory [41] has been proposed by Bulun and colleagues [1]. Briefly, aberrant endometrium with a low level of aromatase expression owing to a possible genetic abnormality is shed into the peritoneal cavity at the time of menstruation. These cells cause subclinical pelvic inflammation and then get some boost from cytokines and PGE-2 derived from the normal immunological scavenger cells, which leads to local estrogen production via an exaggerated feedback circle [6]. The lack of estrogen-neutralizing enzyme (17β-HSD-2), gene-suppressor cofactors and progesterone receptor B prevents the elimination of excess estrogen. Consequently, the survival of endometrial cells leading to the development of endometriosis is supported by estrogen-dependent cell proliferation, together with additional supportive mechanisms such as defective immune functions and/or aberrant expression of other genes.
Concept of estrogen inhibition
The relationship between estrogen and endometriosis seem to be pivotal in the development of the disease. Endometriosis has two main symptoms, chronic pain and infertility, which are caused by endometriotic lesions, adhesions and inflammation in the peritoneal cavity. Current medical treatment aims at altering the menstrual cycle by inducing a state of pseudopregnancy pseudo-menopause or chronic anovulation [6] to inhibit ectopic endometrial growth [42].
There are two main ideas to influence estrogen and its effect on endometriosis. One way is to suppress the production of estrogen by inhibiting its synthetic and regulatory pathway. The other way is to influence estrogen receptors to block estrogen-dependent gene expression.
Estrogen synthesis inhibition
The central repression of estrogen production is achieved by using gonadotropin-releasing hormone (GnRH) analogues that block the synthesis in the ovary by downregulating the pituitary–hypothalamic unit. The treatment results in an approximately 50% reduction in symptoms, with a short-term pain reduction, but in the long-term, pain recurrence can be observed in up to 75% of the cases [43]. GnRH analogues also have considerable side effects, including the loss of bone mass [44].
Recurrence of endometriosis can be partially explained by estrogen synthesis in the adipose tissue and by low-level autocrine estrogen production in the endometriotic cells, which, in a positive-feedback loop, could increase the final estrogen level as explained earlier [3]. Since extra-ovarian estrogen production is not under hormonal control, other sites for inhibition should be considered to aim at this estrogen source.
Aromatase inhibitors
Suppression of extra-ovarian estrogen production in endometriotic lesions and fat tissue could mean a potential treatment of endometriosis. Since aromatase, the key enzyme in estrogen synthesis in ovary, adipose tissue or endometriotic tissue, is encoded by one single gene (the P450arom gene), the inhibition of this gene or its production can cause an effective suppression of estrogen production in all sources [45].
The first generation of aromatase inhibitors (aminoglutethimide and testolactone) [46] was used to block estrogen synthesis in postmenopausal women with breast cancer [47]. The blocking effect was not strictly specific: aminoglutethimide inhibited many other enzymes in steroid biosynthesis, resulted in a medical adrenalectomy [48] and led to important side effects such as lethargy, rashes, nausea and fever [49]. Furthermore, the inhibition was not complete [50].
The second generation of aromatase inhibitors, such as formestane, proved to be effective in the treatment of breast cancer in post-menopuasal women and showed a higher specificity for aromatase, with fewer side effects [51]. These second-generation aromatase inhibitors are defined as the type I group and are mainly steroidal inhibitors [52]. They are derivatives of androstenedione, capable of binding to aromatase as a substrate analogue, inhibiting the conversion to estrogen [53].
The third type of aromatase inhibitors (anastrozole, letrozole, exemestane and vorozole) are 1000- to 10,000-times more potent than the original aminoglutethimide [54]. These compounds are more specific for the aromatase enzyme and produce fewer side effects (headache, nausea and diarrhea). Hot flushes are infrequent [55] and the inhibitory effect is longer; therefore, daily administration is not necessary [56]. These third-generation aromatase inhibitors belong to the type II group of nonsteroidal inhibitors. Type II inhibitors act via binding to P450arom. Aromatase is a member of the cytochrome oxidase family, which contains a heme prosthetic group in covalent association with the protein catalytic part to provide the electron exchange for the redox reaction. Type II inhibitors block this reaction by binding to the heme part [57]. As a physiological consequence, both anastrozole and letrozole cause apoptosis in cultured eutopic endometrial cells [58].
Aromatase inhibitors were first developed for breast cancer treatment [59] and have only recently been considered for the treatment of endometriosis [60]. In one case report, administration of anastrozole successfully relieved pain, caused lesion regression, led to a reduction in estradiol levels in the peripheral circulation, and mRNA levels for aromatase became undetectable in the endometriotic lesion [60]. A number of other publications have been published with respect to the treatment of endometriosis with aromatase inhibitors, as presented in Table 2. Although treatment with aromatase inhibitors has shown promising results, a concomitant treatment is needed in premenopausal women, for several reasons (Table 2). During treatment with aromatase inhibitors, substantial levels of estrogen are still present since the dose used is adequate to shut down peripheral but not ovarian estrogen synthesis. Indeed, lower peripheral estrogen levels following inhibition of extra-ovarian estradiol production stimulate, via a negative-feedback system, the pituitary secretion of luteinizing hormone and follicle-stimulating hormone, resulting in ovarian follicular development and ovarian estrogen production [2,61]. In animal models, only a very high dose of aromatase inhibitor results in a complete block of ovarian estrogen production [62]. Aromatase inhibitors alone are also ineffective in premenopausal women with breast cancer [63]. Therefore, combined treatments of aromatase inhibitors with other hormonal medication (GnRH agonists, progestins, oral contraceptives, and so on) suppressing ovarian estrogen production have been evaluated in women with endometriosis, as shown in Table 2. However, most of these studies are case reports or uncontrolled studies. In the only published randomized trial, a combined treatment with GnRH agonist and aromatase inhibitor therapy was compared with a treatment with GnRH agonist (goserelin) alone in premenopausal women with endometriosis. The combined treatment resulted in a better improvement of pain scores (54.7 vs 10%) and a lower recurrence rate (7.5 vs 35%) than treatment with GnRH agonist only. Patients who received combined treatment had a nonsignificant decrease in bone density [64]. In another study, vaginal administration of anastrazole (0.25 mg) was used in ten premenopausal patients with histologically confirmed rectovaginal endometriosis who were resistant to conventional therapy [65]. Cotreatment with daily calcium and cholecalciferol was provided to prevent loss of bone density. However, a regression in lesion size was observed in only 30% of cases and pain scores were unchanged, possibly owing to the inadequate dose of anastrozole [65]. In summary, the therapeutic effect of aromatase inhibitors without hormonal cotreatment is unknown in women with endometriosis. When premenopausal women with endometriosis are treated with aromatase inhibitors, cotreatment with other hormones is needed to inhibit ovarian stimulation. Preliminary evidence suggests that combined treatment with luteinizing-hormone-releasing hormone analogues and aromatase inhibitors may be superior to medical treatment with luteinizing-hormone-releasing hormone analogues only.
List of aromatase therapies in endometriosis treatment.
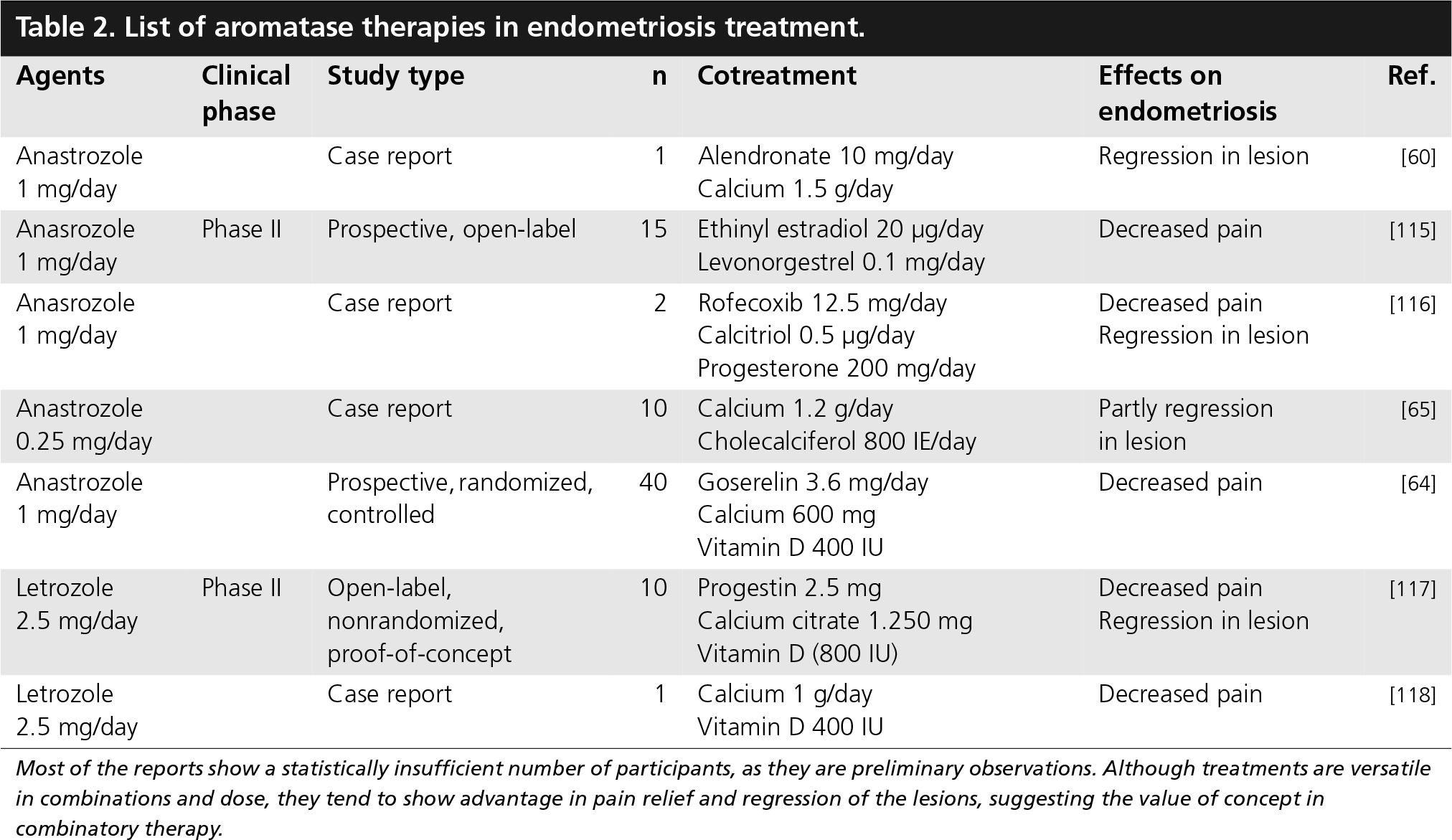
Most of the reports show a statistically insufficient number of participants, as they are preliminary observations. Although treatments are versatile in combinations and dose, they tend to show advantage in pain relief and regression of the lesions, suggesting the value of concept in combinatory therapy.
COX-2 inhibitors
Inhibition of COX-2 synthesis can reduce PGE-2 synthesis and estrogen production, and could support treatment of endometriosis with aromatase inhibitors [30]. NSAIDs, such as aspirin, ibuprofen and diclofenac, have an inhibitory effect on COX-2 and have been used in the treatment of abdominal pain or dysmenorrhea with variable success, which can possibly be explained by their unspecific inhibition of COX-2 [30]. Novel selective inhibitors of COX-2 such as rofecoxib, valdecoxib and celecoxib target intracellular COX-2 and appear to have less gastrointestinal side effects than the NSAIDs [66–68].
In a rat model with induced endometriosis, celecoxib was effective in the prevention of ectopic implants [33]. In a clinical study, treatment of women with minimal-to-mild endometriosis with rofecoxib resulted in significant pain reduction [69]. Unfortunately, rofecoxib and valdecoxib have been withdrawn from the market owing to severe side effects.
Selective estrogen-receptor modulators
Except for raloxifene, which has a specific binding site on target DNA (raloxifene responsive element) [70], selective estrogen-receptor modulators (SERMs) interact with estrogen receptors and block the hormonal signaling pathway. Although SERMs have been proposed in the treatment of endometriosis [55,71], no clinical study is known to have been performed to evaluate their safety and efficacy in patients with endometriosis so far.
The SERMs are polycyclic aromatic compounds with a different chemical structure compared with estrogen, which are capable of binding to estrogen receptors. The SERMs are double-faced agents, having distinct effects on different tissues. In breast cancer they act as estrogen antagonists, while in the bone and on the vasculature they display their agonist activity [72,73]. All SERMs bind to the estrogen receptor and sterically change its conformation, but each SERM acts in a distinct way [74–76]. This unique conformation calls forth the feature allowing the receptor to cooperate with different coregulators (e.g., SRC-1, SRC-2 and SRC-3/AIB1), resulting in different biological outcomes [77,78]. The presence or relative abundance of these coregulators and the availability of regulatory sequences are tissue specific and depend on other factors affecting the cell, such as cytokines [79].
Tamoxifen was the first clinically relevant SERM in breast cancer treatment that obtained US FDA approval in 1999, after it had been proven to be efficient in reducing the chances of developing the disease by 50% in high-risk pre-and post-menopausal women [80,81]. Tamoxifen proved to be effective in reducing breast cancer risk on a long-term basis (7 years) by 49% in a randomly selected patient group [82]. In another study, the same agent was found to reduce the risk of invasive breast cancer by 66% in elderly women with osteoporosis during 8 years of follow-up [83], and was also protective against osteoporosis [84]. Tamoxifen is carcinogenic in rats [85,86] and may cause cancer in the submucosal layer of the human uterus [87], but this risk is reduced in women owing to the liver metabolism of tamoxifen [88]. According to a Phase III trial in women with breast cancer, another aromatase inhibitor, letrozole, had fewer side effects (less intense weight gain, dyspnea, thromboembolic events and vaginal bleeding) than tamoxifen [89].
Executive summary
Endometriosis is an estrogen-dependent disease and an endometrial lesion can produce its own estrogen owing to increased expression of aromatase, and lack of repression proteins. However, aromatase expression/activity can be very low or absent in a considerable number of endometriosis lesions.
When premenopausal women with endometriosis are treated with aromatase inhibitors, cotreatment with other hormones is needed to inhibit ovarian stimulation. The therapeutic effect of aromatase inhibitors without hormonal cotreatment is unknown in women with endometriosis.
COX-2 inhibitors can decrease estrogen production in endometriotic lesions, but their clinical effect in women with endometriosis is unknown, and considerable concern exists regarding their side effects.
Selective estrogen-receptor modulators are potent in decreasing pain and regression of endometriosis lesions in animal models, but have not yet been tested in a clinical environment.
At present, it is too early to conclude that aromatase inhibitors, COX-2 inhibitors or selective estrogen-receptor modulators really present any added value compared with the existing drugs that can be used to achieve hormonal suppression in the medical treatment of endometriosis.
Raloxifene (formerly keoxifene) was developed in the early 1980s as a breast cancer drug, but was less effective than tamoxifen and was not developed further. Later, when tamoxifen turned out to be carcinogenic, raloxifene was studied again, since it does not have carcinogenic effects [90]. Women with increased cardiovascular risk treated with raloxifene are reported to have a reduced incidence of cardiovascular events [91]. It is hypothesized that raloxifene may exert this beneficial effect through nitric oxide release [92]. In vitro exposure of endothelial cells to raloxifene can increase mRNA and protein expression of COX genes that are involved in prostaglandin and prostacyclin synthesis, and this activation is not inhibited by blocking estrogen receptors, suggesting the existence of a yet undiscovered estrogen-receptor-independent mechanism [93].
Toremifene is a relatively new SERM with an action and side effects similar to tamoxifen [94], but with less carcinogenic potential. It has been used successfully in the treatment of cyclical mastalgia with a pain reduction of 77% compared with 35% in placebo-treated control patients [95].
The complex action of raloxifen and other SERMs requires a more careful evaluation, especially regarding possible side effects. The development of SERMs is ongoing. Several new compounds are being evaluated in animal studies, including arzoxifene (LY 353,381·HCl) [96,97], a long-acting raloxifene analogue that protects against bone loss, reduces serum cholesterol levels in rats [96,98], and may provide better results in the treatment of induced mammary cancer than raloxifene [99]. Another compound, EM-652 [loo], seems to have similar positive properties [101]. The compound GW-5638, a tamoxifen analogue, is a total estrogen-receptor antagonist with minimal uterotropic activity [102], whereas molecule SP500263 is a structurally unrelated new SERM [103] with effects comparable with the previously mentioned drugs in the murine breast cancer xenograft model [104].
The value of SERMs in the treatment of endometriosis has been tested in animal models. In rats, raloxifene was tested in various doses (1, 3 or 10 mg/kg) to assess its effect on the regression of endometriotic lesions. Raloxifene was effective in reducing the size of the implanted endometrium only at a 10 mg/kg dose [105]. In conclusion, SERMs are promising drugs, but require more study in the context of endometriosis. The fact that SERMs have different effects in different tissue types makes them interesting agents for further endometriosis research, for example, blocking estrogen-mediated growth of endometriotic lesions and at the same time supporting bone-mass remediation.
Future perspective
Estrogen-pathway inhibition is not the only promising concept in endometriosis therapy There are many other emerging alternatives for hormonal therapy in women with endometriosis, such as levonorgestrel-releasing intrauterine system or selective progesterone-receptor modulators. Furthermore, there is increasing interest in developing nonhormonal medical treatment for endometriosis [106].
Footnotes
The authors have no relevant financial interests, including employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending or royalties related to this manuscript.