
Abstract
Synaptic function is critical for the brain to process experiences dictated by the environment requiring change over the lifetime of the organism. Experience-driven adaptation requires that receptors, signal transduction pathways, transcription and translational mechanisms within neurons respond rapidly over its lifetime. Adaptive responses communicated through the rapid firing of neurons are dependent upon the integrity and function of synapses. These rapid responses via adaptation underlie the organism's ability to perceive, learn, remember, calculate and plan. Glutamate, the endogenous neurotransmitter required for physiological excitation in the brain, is critically involved in neuronal adaptive responses and in the pathophysiology of neurodegenerative disorders. Using neuronal experimental systems, we will discuss how compounds with low dose effects mediated via glutamate receptors can result either in a neuroprotective or neurotoxic response. Because the brain has evolved to respond rapidly to environmental cues, exposure of neurons to stressful stimuli can result in a pivotal response toward either synaptic adaptation or dysfunction and neuronal cell death. Understanding how neurons adapt to stressful stimuli will provide important clues toward the development of strategies to protect the brain against neurodegeneration.
INTRODUCTION
Brain-derived neurotrophic factor is widely distributed in brain and exhibits early immediate gene kinetics. BDNF exerts diverse function in brain including neuronal cell maintenance, survival, neurogenesis, maturation and plasticity (Kaplan and Miller, 2000; Miller and Kaplan, 2001; Lee et al., 2002; Blondeau et al., 2009; Greenberg et al., 2009). BDNF plays a critical role in long-term potentiation (LTP) presynaptically (Xu et al., 2000) and postsynaptically (Kovalchuk et al., 2002) and in the maintenance of high frequency stimulation-induced LTP in the adult rat (Gärtner and Staiger, 1992). Many of these effects are mediated by the high affinity receptor TrkB, which are localized on cell bodies and dendrites as well as postsynaptically (Yan et al., 1997).
The human BDNF gene spans more than 70 kb and is composed of seven 5’ exons that are differentially spliced to a single 3’ terminal exon (Liu et al., 2005). Each 5’ exon is transcribed from a unique promoter. This complex transcriptional regulation leads to the creation of at least three pre-pro-BDNF isoforms, the function of which is not completely understood (Marini et al., 2004; Liu et al., 2005). This overall gene architecture is highly conserved in mammals. In rat, four promoters from BDNF were characterized by Timmusk et al. (1993). The well-known BDNF 5’ flanking region of exon 4 (previously known as exon 3 [Timmusk et al., 1993]) is activated by membrane depolarization in cultured cortical and hippocampal neurons. Other activators of BDNF exon 4–specific transcription have been described, including glutamate via NMDA receptors (Lipsky et al., 2001), kainate via non-NMDA receptors (Nakayama et al., 1994), and dopamine (Fang et al., 2003), with at least three transcription start sites mapped within an 80-bp region of DNA known to contain activation-dependent start sites (Timmusk et al., 1993; Nakayama et al., 1994; Tao et al., 1998). Expression patterns of exon-specific BDNF transcripts in the brain are developmentally regulated, but the mechanisms remain largely unknown. However, activity-dependent neuronal plasticity resulting in the modulation of the synaptic strength of existing synaptic connections and in the formation of new synaptic contacts, are regulated in part by BDNF (Thoenen, 1995; Katz and Shatz, 1996; Lu and Figurov, 1997; Chao, 2003). These functions may require the synthesis of exon-specific BDNF transcripts. Thus, differential BDNF promoter use may be targeted by different signal transduction pathways to promote the diverse functions of BDNF (Timmusk et al., 1994; McAllister et al., 1995; Ringstedt et al., 1998; Kafitz et al., 1999; Poo, 2001; Pattabiraman et al., 2005; Wang et al., 1995).
Transcription factors regulate gene expression by binding to specific promoter sites most frequently encountered preceding the protein coding region of a gene. Our group discovered that basic helix loop helix B2 (BHLHB2) regulates promoter IV of the bdnf gene, a major promoter that drives activity-dependent BDNF expression (Jiang et al., 2008). BHLHB2 is a transcriptional repressor and a nuclear protein (Jiang et al., 2008). One of the most interesting features of BHLHB2 is the fact that this protein is induced by hypoxia in cancer cell lines (Ivanova et al., 2001). Thus, regulation of promoter IV of the bdnf gene by BHLHB2 may occur under hypoxic as well as normoxic conditions and differences in oxygen level may dramatically affect exon 4-specific-mediated BDNF expression and possibly function.
Neuronal function is critically dependent upon receptors and signal transduction, which impact transcriptional and translational processes that are influenced by genetic and environmental (epigenetic) mechanisms. Dysfunction in any one or a combination of these molecular processes can lead to neuronal cell death by increasing the vulnerability of neurons to ion fluxes, increased energy requirement via oxidative stress, a reduction in existing intrinsic survival pathways, DNA damage, and/or alterations in chromatin remodeling. Environmental factors may also affect glutamate receptor-mediated neurotransmission. The outbreak of encephalopathy due to eating mussels contaminated with the toxin domoic acid from the diatom Nitzschia pungens (Perl et al., 1990) is an example of how food consumed by humans can affect the brain and specifically alter glutamate receptor function (Debonnel et al. 1989, Tasker et al. 1991, Novelli et al., 1992, Tasker and Strain 1998; Novelli et al., 2005). Domoic acid is a tricarboxylic excitatory amino acid that binds to and activates AMPA as well as kainic acid receptors with low and high affinity respectively (Novelli et al., 2000). Moreover, the human neuropathology caused by domoic acid in the hippocampus is similar to that found in animals injected with kainic acid (Teitelbaum et al. 1990). Attention has been raised recently on the potential risk for human health that other compounds found in oceans may represent. One of the marine compounds most toxic to mammals is Palytoxin, a polyalcohol molecule synthesized by microalgae of the genus Ostreopsis (Moore and Scheuer, 1971; Usami et al., 1995), and represents a human health hazard (Alcala et al., 1988; Onuma et al., 1999). Palytoxin selectively binds to and alter the Na+/K+-ATPase pump (Bottinger et al. 1986, Habermann 1989, Scheiner-Bobis et al. 1994, Artigas and Gadsby, 2003) and recently palytoxin has been shown to facilitate excitotoxicity via non-NMDA receptors at subtoxic concentrations (Pérez-Gómez et al. 2010).
In this paper, we investigated whether alterations in oxygen levels may affect BHLHB2 expression in cultured hippocampal neurons and modulate exon 4-specific BDNF mRNA levels, thus linking environmental changes to either neuroprotection or excitotoxicity. Furthermore, we also investigated whether palytoxin, a compound synthesized in the marine environment, activates intracellular pathways leading to neuroprotection as opposed to excitotoxicity, similar to the results found for the activation of glutamate receptor subtypes (Marini and Novelli, 1991; Marini and Paul, 1992; Wu et al., 2004).
METHODS
Preparation of hippocampal neurons.
Hippocampi were prepared from embryonic day 20 fetuses obtained from Sprague-Dawley dams (Taconic Farms, Germantown, NY). Tissue from 8–10 fetuses was dissociated by mild trypsinization and trituration as described (Cheng et al., 1995; Jiang et al., 2005; Jiang et al., 2008). Hippocampal cells were then plated onto poly-D-lysine-coated culture dishes. Hippocampal neurons were maintained under serum-free conditions throughout the cultivation period. One-fifth of the medium was removed and replaced with fresh medium every third day to replenish nutrients. All experimental treatments were performed on neurons on day in vitro (DIV) 7–8, which contained 90–95% neurons. Hippocampal neurons cultured in serum-free medium have been extensively characterized using neuronal and glial antibodies (Perry et al., 2003; Fu et al., 2002 and references therein; Jiang et al., 2008). This research was conducted according to the principles set forth in the National Institutes of Health (NIH) Publication No. 85–23, Guide for the Care and Use of Laboratory Animals, and the Animal Welfare Act of 1986, as amended. Every effort was made to minimize the number of animals.
Cerebellar neuron cultures and neurotoxicology.
Primary cultures of rat cerebellar neurons were prepared as previously described (Novelli et al., 1988). Cerebellar neuronal survival was maintained for more than 40 days in culture by replenishing the culture medium with glucose every four days and replacing evaporative losses with sterile water. The animal procedures used were in accordance with the protocols approved by the Institutional Animal Care and Use Committee of the University of Oviedo, Oviedo, Spain. Neurons were used between 14 and 20 days in culture. Drugs were added into the growth medium or the incubation buffer at the indicated concentrations, and neuronal cultures were observed for signs of neurotoxicity thereafter by phase contrast microscopy.
cGMP determination.
Intracellular cGMP concentration was determined as previously reported (Novelli and Henneberry, 1987). Briefly, cultures were washed with 1 ml prewarmed (37°C) incubation buffer containing (in mM): 154 NaCl, 5.6 KCl, 5.6 glucose, 8.6 HEPES, 1 MgCl2, 2.3 CaCl2, pH 7.4. Dishes were incubated at 37°C for 10 min. with 1 ml new incubation buffer and for an additional 20 min. with a second 1 ml new incubation buffer. Drugs were added at the end of the 20 min. incubation period for the indicated times. Incubation was stopped by aspiration of the solution and addition of 1 ml HClO4 (0.4 N). After neutralizing the perchlorate extract, cGMP content was determined by radioimmunoassay (125I). Protein content was determined on the membrane pellet from the same sample as described previously (Novelli and Henneberry, 1987).
Determination of neuronal survival.
To quantify neuronal survival, cerebellar granule cell cultures were stained with fluorescein diacetate and ethidium bromide (Novelli et al., 1988), photographs of three randomly selected culture fields were taken and live (green) and dead (red) neurons were counted. Results are expressed as percentage of live neurons. Total number of neurons per dish was calculated considering the ratio between the area of the dish and the area of the pictures (∼3000).
Drug treatments.
A subtoxic concentration of palytoxin (PTX, 100 pM) was added 3h prior to the addition of a subtoxic concentration of domoic acid (DOM, 5 μM). 6-cyano-7-hydroxy-5-methylisoxazole-4-propionate (CNQX, 15 μM), a non-NMDA receptor antagonist, or MK-801 (1 μM), an NMDA receptor antagonist, was added 5 min prior to DOM. Neuronal survival was quantified 24h after addition of DOM and expressed as the percentage of live neurons.
Preparation of nuclear extracts.
Nuclear extracts were prepared according to the manufacturer's instructions (Active Motif Inc., Carlsbad, CA) with minor modifications. Primary hippocampal neurons at day in vitro (DIV) 8 (7–8 × 105 neurons) were washed with 0.7 ml ice-cold PBS plus Phosphatase Inhibitors (Active Motif, Carlsbad, CA) for each 35 mm dish and replaced with 0.5 ml ice-cold PBS/Phosphatase Inhibitor. Neurons were removed from the dish by gentle scraping and the cells from three dishes pooled. The suspension was centrifuged for 2 min at 100 x g, 10 min, 4°C. The cell pellet was gently resuspended in 250 μl Hypotonic Buffer (Active Motif, Carlsbad, CA) and the suspension incubated at 4° C for 15 min followed by the addition of Triton-x100 (12.5 μl) to each tube followed by brief vigorous shaking by vortex (10 sec at the highest setting). The suspension in each tube was centrifuged at 14,000 × g for 1 min in a microcentrifuge pre-cooled at 4°C. The supernatant (cytoplasmic fraction) from each tube was transferred into a clean pre-chilled microcentrifuge tube. The remaining nuclear pellet was then resuspended in 50 μl Complete Lysis Buffer (Active Motif, Carlsbad, CA) followed by vigorous shaking on a Vortex for 10 sec at the highest setting. The suspension in each microfuge tube was incubated for 30 min on a rocking platform at 4°C followed by vigorous shaking by vortex for 30 sec at the highest setting. The samples were centrifuged for 10 min at 14,000 × g at 4°C. The supernatants from each microfuge tube (nuclear fraction) were placed in a pre-chilled microcentrifuge tube and stored at −80°C. Protein concentration was determined by the Bradford assay (BioRad, Hercules, CA).
Western blot analysis.
Western blot analyses were performed using forty μg of nuclear extract protein. Proteins were first fractionated on 10% SDS-PAGE gels and transferred to nitrocellulose membranes (BioRad, Hercules, CA). The blots were then blocked with Tris-buffered saline containing 0.2% Tween 20 (TBST) and 5% non-fat dry milk followed by three washes in TBST. The blots were incubated with anti-BHLHB2 antibody (1:200, Aviva Systems Biology, San Diego, CA) or anti-TATA box binding protein (TBP, 1:1000, Sigma-Aldrich, St. Louis, MO) overnight at 4°C. TBP was used as a nuclear protein marker to ensure equal loading of protein. After three washes with TBST containing 5% non-fat dry milk, membranes were incubated with the appropriate secondary antibody linked to horseradish peroxidase for 1h. Immunoreactive bands were visualized using the enhanced chemiluminescence detection system according to the manufacturer's recommendations (SuperSignal West Femto Maximum Sensitivity Substrate, Thermo Scientific) [Wu et al., 2004]. Immunoactive bands were quantified by image analysis using a scanner (Hewlett-Packard) and Image J analysis software (NIH, Bethesda, MD). The fold of induction is the quotient of the density of the band divided by the density of the band at time zero and normalized to TBP density. All experiments were performed in triplicate using two different neuronal preparations.
Oxygen deprivation/hypoxic conditions.
Hippocampal cultures were seeded in 35 mm Nunc® culture dishes as described above. At the time of the experiment, serum-free medium containing all nutrients (Jiang et al., 2008) was exchanged for deoxygenated (60 min nitrogen aeration) serum-free medium (Skaper et al., 1991). Cultures for hypoxia time-course experiments were placed in a humidified incubator containing 1% oxygen/5% CO2/94% N2 whereas control cultures were harvested immediately after exposure to deoxygenated culture medium.
Quantitative real-time PCR. Hippocampal primary neurons from rat cultured in 35 mm Nunc® culture dishes were lysed directly in the culture dish by removing the culture medium and adding 0.5ml RNA STAT-60 (Qiagen, Germantown, MD). The cell lysates were transferred to new sterile Eppendorf tubes and allowed to stand at room temperature for 5 min. The samples were placed at −80°C for 1 h, followed by addition of 0.1ml chloroform per 1 ml of RNA-STAT-60, and the samples were shaken vigorously for 15 sec and allowed to stay at room temperature for 2–3 min. The cell lysates were centrifuged at 12,000 × g for 15 min at 4°C. The upper aqueous phase was placed in a fresh Eppendorf tube and absolute ethanol added such that the final volume was 53% ethanol v/v. Each sample was placed on an RNeasy spin column (Qiagen, Germantown, MD) and centrifuged for 15 sec at 8000 × g. The column was washed first with 700 μl Buffer RW1 then twice of 500 μl Buffer RPE for spun for 1 min at 8000 × g. RNase-free water (30–50 μl) was added directly to the spin column membrane and centrifuged for 1 min at 8000 × g to elute the RNA. The purified RNA was subjected to RNase-free DNase digestion (Ambion, Austin, TX) and re-extracted prior to being stored at −20°C. The first strand cDNA synthesis was prepared using Cloned AMV First –strand cDNA Synthesis kit (Invitrogen, Carlsbad, CA) using 120 ng of total RNA for each reaction. Levels of specific mRNAs were determined using realtime fluorescence 5′-nuclease (TaqMan) assays. The TaqMan (Applied Biosystems, Foster City, CA) assay using fluorogenic detection probes to BHLHB2, exon 4 and exon 7 of BDNF were performed as described previously (Jiang et al., 2008). Levels of specific mRNA from hippocampal tissue were normalized to endogenous Gapdh mRNA levels (rodent Gapdh mRNA assay; Applied Biosystems, Foster City, CA). Fold differences of specific mRNAs relative to control were calculated by an algorithm that determines the threshold cycle (Ct) when an increase in reporter fluorescence above a baseline signal is first detected during PCR. The sequence detection software generates a calibration curve of Ct versus amount of reference cDNA and then determines unknown amounts by interpolation. Real-time fluorescence detection was performed using a 7700 Sequence Detector (Applied Biosystems, Foster City, CA). RT-PCR assays were performed in duplicate or triplicate from two different neuronal preparations.
Statistical analysis.
All values are presented as mean ± SD or percent of control (untreated). The Student t test was used for analysis; p < 0.05 is considered significant.
Materials. 6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX), AMPA, amino-phosphono-valeric acid (APV) and domoic acid were purchased from Tocris Cookson (Bristol, UK). NS102 was a gift from Dr. J. Drejer (Neurosearch, Glostrup, Denmark), and palytoxin was obtained from Calbiochem (Merck, UK). All other chemicals were obtained from commercial sources.
RESULTS
Hypoxia increases BHLHB2 protein expression in hippocampal neurons.
BHLHB2 is a protein expressed in brain (Rossner et al., 1997) and a hypoxia-inducible gene in tumor cell lines (Ivanova et al., 2001) and cancer cells (Turley et al., 2004). However, the effect of hypoxia on this protein in primary neurons is unknown. We first investigated the effect of oxygen deprivation/hypoxia on BHLHB2 protein expression by exposing primary rat hippocampal neurons to hypoxic conditions (1% oxygen). BHLHB2 protein expression is low following brief exposure of the neurons to deoxygenated serum-free medium (control). We performed a standard time course to determine whether BHLHB2 protein levels increased under hypoxic conditions. Hippocampal neurons were subjected to oxygen deprivation/hypoxia for various times (see Methods), conditions that do not affect their viability throughout the duration of the experiment (data not shown). BHLHB2 protein expression was quantified and compared to the TATA box binding protein, a nuclear protein that remains constant over time. BHLHB2 protein expression increases and peaks at 3h followed by a return to baseline levels within 24h (Figure 1).
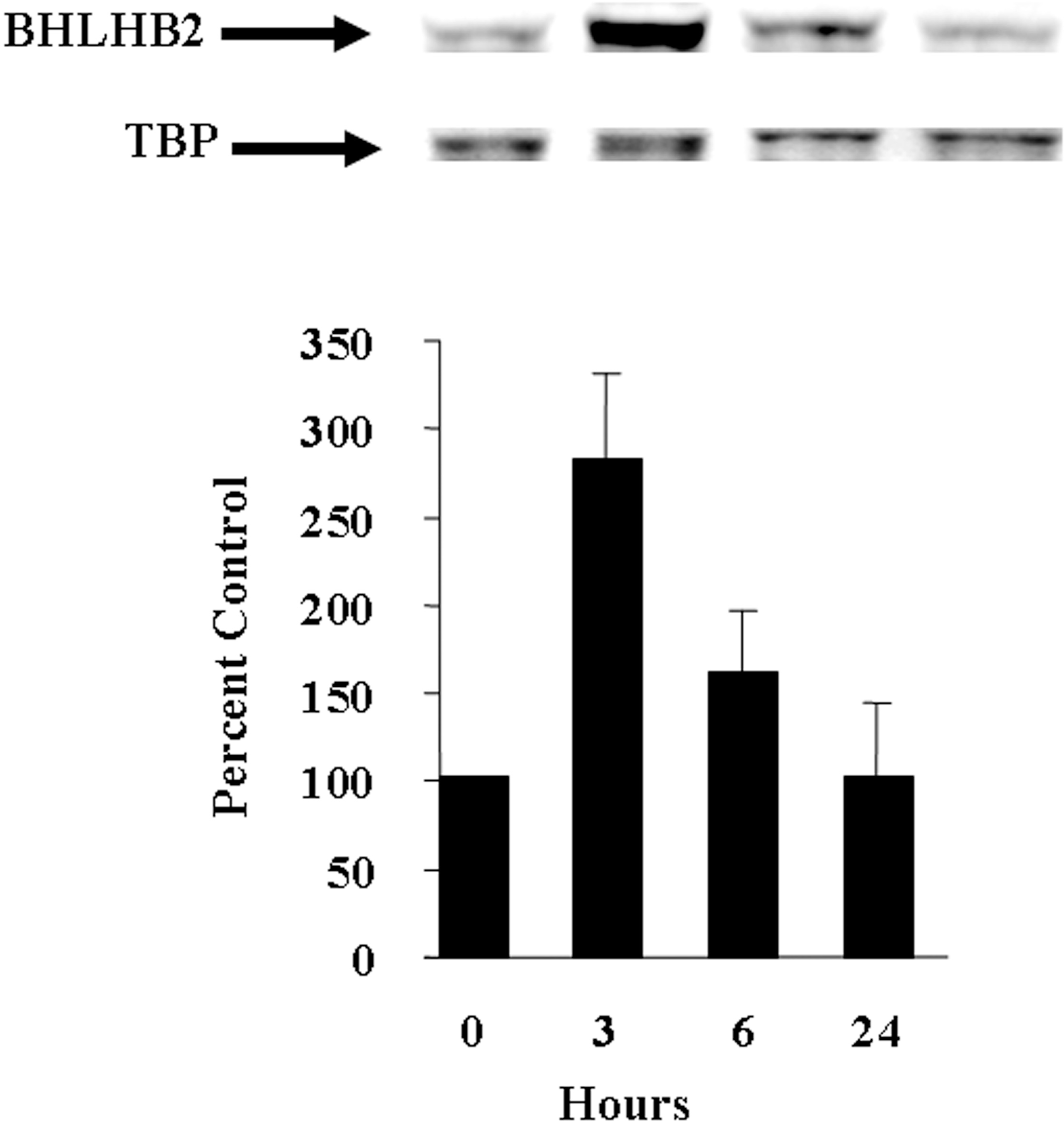
Hypoxia increases BHLHB2 protein expression in cultured hippocampal neurons. Cultured hippocampal neurons were exposed to hypoxia in a humidified incubator for various times. At the indicated time, the neurons were harvested, nuclear extracts prepared followed by a standard western blot to detect BHLHB2 and TBP. Results are expressed as the mean ± SD (n = 6).
Our recent study showed that BHLHB2 represses promoter IV activity of the BDNF gene under normal (95% air/5% CO2) conditions (Jiang et al., 2008). To determine the transcription expression levels of BHLHB2 and exon-specific BDNF levels under hypoxic conditions, cultured rat hippocampal neurons were subjected to oxygen deprivation/hypoxic conditions for various times; BHLHB2 and BDNF exon 4- and exon 7-specific mRNA levels were quantified by real-time PCR. BHLHB2 mRNA levels increase and reach a peak of 5.5-fold at 6h while exon 4-specific BDNF transcripts increase rapidly to 5.6-fold at 2h but then decrease steadily over the remainder of the experiment. Coincidently, the decline in exon 4-specific BDNF transcripts occurs when BHLHB2 mRNA levels increase over the same time course. Exon 7-specific BDNF transcripts, the terminal exon in rat that synthesizes mature BDNF protein (Marini et al., 2004) rises more slowly reaching a maximum of 2.6-fold at 5h and then decreases over the next three hours during the same time that BHLHB2 mRNA levels are maximal (Figure 2).
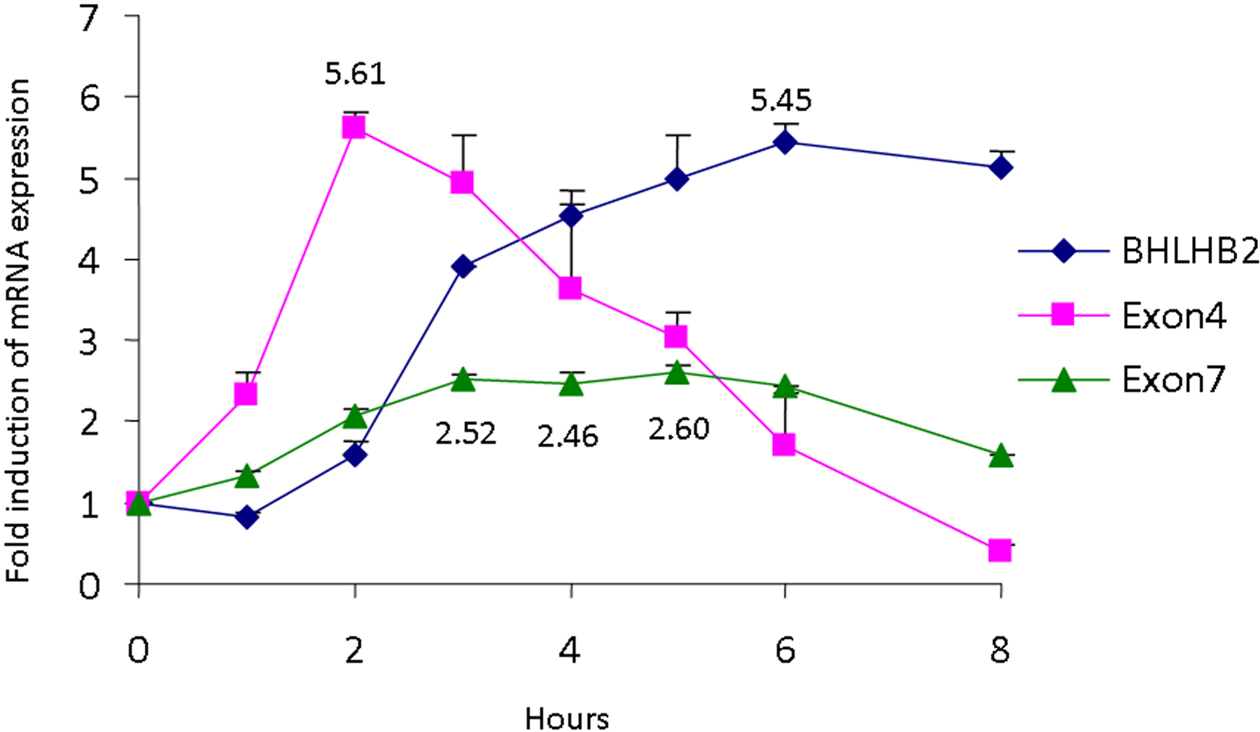
Hypoxia-induced changes in exon 4- and exon 7-specific BDNF and BHLHB2 mRNA levels in cultured hippocampal neurons. Cultured rat hippocampal neurons were exposed to hypoxic conditions for various times. At the indicated time, neurons were harvested and total RNA isolated. Exon 4- and exon 7-specific BDNF and BHLHB2 mRNA levels were quantified by the 5’ nuclease assay as described elsewhere (Jiang et al., 2008). The fluorescence of the mean of each experiment was compared with glyceraldehydes phosphate dehydrogenase (GAPDH) mRNA and is expressed as relative fluorescence. Each experiment was performed in triplicate using two different preparations of neurons (n = 6).
Palytoxin evokes neuroprotection or excitotoxicity in cultured cerebellar granule cells.
Palytoxin modulation of non-NMDA glutamate receptors activated by domoic acid is one means by which chemicals synthesized by oceanic plants affect neuronal function. Thus, exposure of cultured cerebellar neurons to domoic acid (DOM) elicits a potent concentration-dependent increase in intracellular cGMP with an EC50 of ≈ 4μM and an EC100 of 7.5μM (Figure 3a). The increase in cGMP elicited by DOM (10 μM) is fully blocked by the non-NMDA receptor antagonist CNQX but not by the NMDA receptor antagonists APV or MK-801 (Figure 3b). Furthermore, DOM stimulation of cGMP can be reduced by preventing activation of either AMPA or kainate receptors with either the desensitizing agonist AMPA or with NS-102, an antagonist of kainate receptors (Figure 3b) (Gouaux 2004, Lerma et al. 1993). A twenty four hour exposure of cultured cerebellar neurons to 5 μM DOM fails to elicit excitotoxicity (Figure 4). However, when cultures were pretreated with a subtoxic concentration of palytoxin (PTX, 100 pM), the exposure to DOM (5μM) results in a dramatic reduction in neuronal survival after 24h that can be prevented by the prior addition of CNQX but not MK-801 (Figure 4). The facilitation of DOM toxicity by palytoxin is dependent upon the length of time the cultures are pretreated with this marine compound. A significant facilitation is obtained after 5 min, and a maximal facilitation is achieved after a 5h pretreatment time. Longer pretreatment times with palytoxin results in a progressive reduction of excitotoxic facilitation by DOM and disappears after a 48h palytoxin pretreatment time (Figure 5). To exclude that this unexpected effect could be due to the inactivation or degradation of the marine compound, palytoxin (100 pM) was added to the culture medium of a group of neuronal cultures for 48h. Then culture medium from naïve sister cultures was removed and replaced with either that of 48h palytoxin-treated cultures (48h PTX-containing 48h-CM) or with culture medium from untreated cultures to which palytoxin was freshly added (PTX). Three hours later, either a vehicle solution or a subtoxic concentration of domoic acid (5 μM) was added to the medium of each culture group and neuronal survival was quantified 24 hours later. The addition of DOM in neuronal cultures where the culture medium was replaced with 48h PTX-containing 48h-CM results in a reduction in neuronal survival identical to that observed in cultures where palytoxin was freshly added whereas exchanging and replacing sister culture medium from naïve cultures followed by the addition of only DOM did not affect neuronal viability (Figure 6). These results support the idea that effects other than degradation or inactivation of palytoxin are responsible for the increase in neuronal survival following non-NMDA receptor stimulation by DOM when neurons are exposed to subtoxic concentrations of palytoxin over prolonged exposure times.
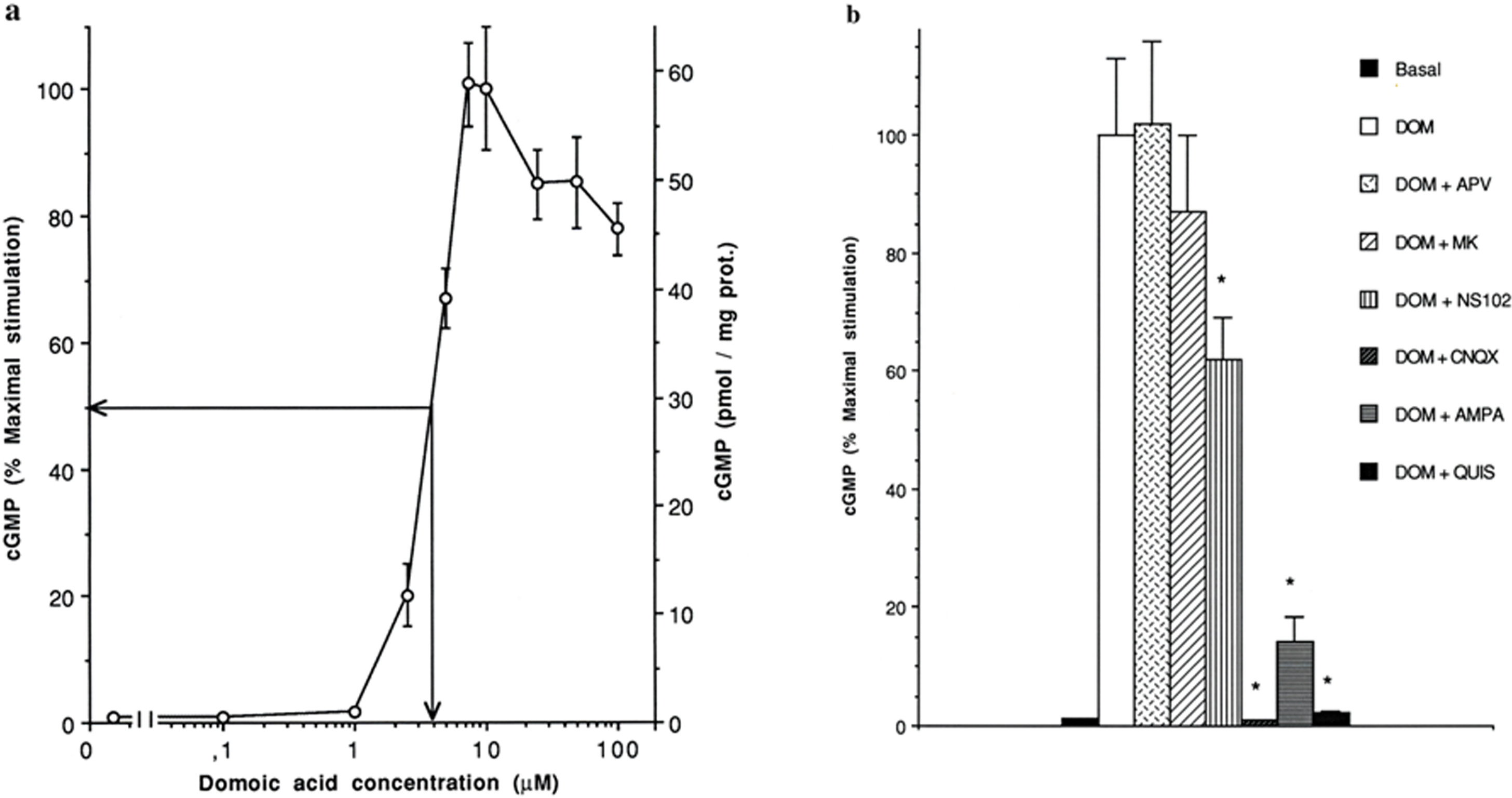
Domoic acid stimulates intracellular cGMP formation in a concentration-dependent manner via non-NMDA receptors. Panel a: Intracellular concentration of cGMP was measured in cerebellar cultures at 12–14 day in culture (DIC) following 1 min stimulation with increasing concentrations of domoic acid (DOM). Intracellular cGMP content was determined by radioimmunoassay. Represented values are the mean ± SD of duplicate values from three independent experiments. Panel b: Pharmacological characterization of cGMP formation following domoic acid stimulation was measured at 12–14 DIC. All drugs were added 10 min before domoic acid (DOM 10 μM) and cultures were exposed to DOM for 1 min. Concentrations were: CNQX 15μM, MK801 (MK) 2μM, APV 1mM, AMPA 200μM, NS102 10μM, Quisqualic acid (QUIS, 100μM). Represented data are the mean ± SD of duplicate values from three independent experiments. *p < 0.01 by student t test vs. DOM alone.

The non-NMDA receptor antagonist, CNQX, blocks domoic-induced neuronal cell death enhanced by palytoxin. A subtoxic concentration of palytoxin (PTX, 100 pM) was added to cultured rat cerebellar granule cells 3h prior to the addition of a subtoxic concentration of domoic acid (DOM, 5 μM), The percentage of viable and degenerating neurons quantified. The addition of domoic acid (5 μM) to neurons previously exposed to palytoxin reduces neuronal survival by 70%. Addition of CNQX, a non-NMDA receptor antagonist, but not with MK-801, an NMDA receptor antagonist, completely blocks the excitotoxic effect. No effect on neuronal survival was observed with domoic acid or palytoxin alone compared with untreated neurons.

Palytoxin enhancement of domoic acid-induced excitotoxicity is biphasic and depends upon the length of exposure time. Cultured rat cerebellar granule cells were treated with a subtoxic concentration of palytoxin (100 pM, open circles) for various times (5 min-48h) prior to the addition of a subtoxic concentration of domoic acid (DOM, 5 μM). Addition of palytoxin alone (100 pM) does not affect neuronal survival (closed circles). Represented data are the mean ± SD of duplicate values from three independent experiments (n = 6).

Palytoxin is not inactivated following long-term incubation with cultured neurons. A subtoxic concentration of palytoxin (PTX, 100 pM) was added to cultured rat cerebellar granule cells for 48h. Palytoxin-containing media (palytoxin-containing 48h-CM [conditioned media]) was transferred to naïve cultures followed by the addition of a subtoxic concentration of domoic acid (DOM, 5 μM) and neuronal survival was quantified 24h later. Addition of medium from control cultures alone (control 48h-CM) to naïve cultures in the presence or absence of a subtoxic concentration of domoic acid (DOM, 5 μM) served as the control. Addition of a subtoxic concentration of palytoxin for 3h followed by the addition of a subtoxic concentration of domoic acid (DOM, 5 μM) freshly added resulted in an excitotoxic response and served as the positive control (n = 6).
DISCUSSION
The results in this paper show that BHLHB2 is regulated by hypoxia in cultured hippocampal neurons. The apparent inverse relationship between BHLHB2 mRNA and exon 4-specific BDNF mRNA levels is an interesting association because BHLHB2 binds to and represses promoter IV of the bdnf gene, a major promoter that drives activity-dependent BDNF expression (Jiang et al., 2008). Additional experiments will determine the role of BHLHB2 in regulating exon 4-specific BDNF mRNA levels in cultured neurons under hypoxic and preconditioning conditions. Taken together, modulation of oxygen is one of many mechanisms responsible for changes in neuronal gene and protein expression activated by ionotropic glutamate receptors. Elucidating the role of BHLHB2 in models of preconditioning (Marini et al., 2008) may contribute to delineating additional neuroprotective pathways and toward more efficacious drugs to protect the brain.
Organisms in the oceans synthesize a vast array of compounds, some of which may alter the function of ionotropic glutamate receptors through synergistic mechanisms that determine cell fate. Palytoxin is a compound synthesized by oceanic microalgae and transforms the Na+/K+ ATPase pump into a non-selective channel permeable to monovalent cations (Scheiner-Bobis et al., 1994). The results presented in this paper show that modulation of the Na+/K+-ATPase activity by palytoxin plays a crucial role in determining neuronal susceptibility to non-NMDA receptor-mediated excitotoxicity, confirming and expanding previous results by Pérez-Gómez et al. (2010). Subtoxic concentrations of palytoxin or domoic acid alone do not affect neuronal survival. Addition of a subtoxic concentration of domoic acid three hours after the addition of a subtoxic concentration of palytoxin results in a rapid excitotoxic response. The excitotoxic effect is mediated by non-NMDA receptors because excitotoxicity is completely blocked by prior treatment with CNQX, a non-NMDA receptor antagonist. It is worth noting that the subtoxic concentration of domoic acid used (5μM) potently stimulates the intracellular formation of the second messenger cGMP, and that the reduction to a similar extent of cGMP stimulation during exposure to a toxic concentration of DOM (10 μM) did not prevent excitotoxicity (Figure 3b and data not shown), suggesting that the pathway leading to the formation of this messenger is not involved in excitotoxicity (Novelli et al. 2005). Curiously, palytoxin does not affect susceptibility to NMDA receptor-mediated excitotoxicity (Figure 3 and Pérez-Gómez et al. 2010). In addition, palytoxin exerts its own dose effect depending upon the incubation time. That is, the longer palytoxin is exposed to the neurons the lower the excitotoxicity following domoic acid. Longer exposure times of palytoxin do not result in the inactivation of the compound because a prompt excitotoxic effect is observed when the palytoxin-containing medium is added to naïve neuronal cultures followed by the addition of domoic acid (Figures 5–6). The mechanism of this differential effect remains elusive but it is possible that cellular adaptation at the level of the Na+/K+-ATPase or downstream factors may be responsible for the reduced effect on excitotoxicity.
Modulation of NMDA receptors or Na+/K+-ATPase may result in alterations in brain function. It has been shown that activation of NMDA receptors is required to protect vulnerable brain regions against hypoxic-ischemic insults in models of preconditioning (Kirino, 2002; Chazot et al., 2002) and against traumatic brain injury (Ikonomidou et al., 2000). Activity-dependent release of BDNF through direct activation of NMDA receptors (Marini et al., 1998) or in a model of memory known to require NMDA receptors (Gartner and Staiger, 2002) may provide important clues as to how low dose effects modulate neuronal function to protect the brain against neuropathological conditions known to involve glutamate as a final common pathway. Thus, activity-dependent processes via NMDA receptors and neurotrophins such as BDNF may be required for neurons to promote their own survival such that in the absence of oxygen and glucose, overactivation of NMDA receptors leads to neuronal cell death. Likewise, a reduction in Na+/K+-ATPase function either through decreases in activity or density is known to occur in hypoxic-ischemic neuronal injury and following the administration of glutamate receptor agonists (Mintorovitch et al., 1994; Lees, 1991; Tavalin et al., 1997). In contrast, an increase in Na+/K+-ATPase is a crucial event in ischemic preconditioning (Tian et al., 2008). The results showing that palytoxin modulates non-NMDA receptor-mediated neuronal cell death may have important implications in neuronal function and dysfunction in the brain. Because palytoxin and domoic acid are seafood contaminants, ingestion of low concentrations (possibly below those actually allowed in seafood) of both toxins may have opposing effects on human health depending upon the amounts and the kinetics of the drugs crossing the blood brain barrier.
In summary, low dose effects may have profound effects on brain function. Delineating the mechanisms of low dose effects in the brain may provide important insights on neuronal function and lead to compounds that enhance efficacy and reduce unwanted side effects.
Footnotes
ACKNOWLEDGMENT
This work was supported by the Defense Threat Reduction Agency, grant numbers CBM.NEURO.01.10.US.019 (AMM), CBM.NEURO.01.10. US. 012 (AMM) and by CICYT, Grants AGL2004-08241-C04-04/ALI, AGL2005-07924-C04-03/ALI and CTQ2008-06754-C04-03/PPQ (AN and MTFS). A. Pérez-Gómez was a fellow of FICYT.