
Abstract
Amifostine [S-2-3-aminopropil amino ethyl phosphorotioic acid], a modulator agent for antineoplastic drugs involved in free radicals generation has given controversial results in cisplatin treated leukocytes in vitro. We have evaluated the amifostine protection over leukocytes in vivo, using comet assay. Groups of five OF1 male mice were given one of three doses of amifostine (56, 105 and 200 mg/Kg) after a cisplatin single injection (10 mg/Kg). Serum malonyldialdehide levels, catalase and superoxide dismutase activity were also evaluated. Amifostine showed significant DNA protection (p< 0.01) at the two lower doses evaluated. Malonyldildehide decreased in all amifostine treatments with respect to cisplatin while antioxidant enzyme activities remained unchanged. However, DNA migration increased with the highest amifostine dose; in fact highest dose of amifostine did no protect damage caused by cisplatin this result have implications on amifostine treatment schedules in clinical practice.
INTRODUCTION
Last decades have witnessed an impressive advance in cancer survival in western countries; that success is the result of a combination of more sensitive diagnostic and staging methods, a better understanding of cancer biology and refinements in radio, inmuno, surgical and chemical treatments. It is widely recognized that introduction of poly-chemotherapy accounted for a significant improvement in survival rates. (Ries et al. 2006) However, oncospecific drugs cause DNA damage and mutation that could be expressed as an increased frequency of second neoplasms among cancer survivors (Kollmannsberger et al. 1999).
The use of cytoprotective drugs in combination with antineoplastic agents has aroused as a possibility to minimize or avoid primary genetic damage. Amifostine [S-2-3-aminopropil amino ethyl phosphorotioic acid] or WR2721, has been introduced as radio protective and also against platinum derived drugs toxicities (Hospers et al 1999). Amifostine protection relies on its different behavior in normal and tumor tissues. In the former, the prodrug is activated through desphosphorilation by membrane bound alkaline phosphatase, resulting in WR1065 and other metabolites thiol-based free radicals scavengers. Whereas in tumoral tissues, physiopathological conditions as hypoxia, low interstitial pH and lower expression of alkaline phosphatase, results in activation rates a hundred times lower than in normal ones. Yuhas (1980). This particular drug metabolism confers a selective protection to non-cancer cells against oxidative damage (Mertsch et al. 1998; Kouvaris et al. 2007).
Cisplatin (cis diamino dichloroplatinum) (Bergström et al. 1999) develops toxicity, especially in kidney, liver and bone marrow (Boulikas and Vougiouka 2003). Also leukocytes's DNA is damaged by cisplatin adduction short after the infusion of this drug (Lieder et al. 2006). Cisplatin could induce intra- and interstrand DNA-DNA cross-links, as well as DNA-protein crosslinks (Wozniak et al. 2004). There are evidences of protection over human leukocytes by amifostine; Pierelli et al. 2004 demonstrated that amifostine pretreatment in vivo protected peripheral blood mononuclear cells from the toxic effect of etoposide, carboplatin and taxotere. WR2721 rescued peripheral blood lymphocyte from apoptotic DNA fragmentation induced by cysplatin, adriamycin, and cyclophosphamide (Provinciali et al. 1999). However, there are controversies upon the capacity of amifostine to protect peripheral blood leukocytes against free radicals, generated by those DNA damaging agents (Littlefield and Hoffmann 1993; Bhattacharya et al. 2001; Campos Nebel et al. 2002; Xunclà et al. 2008).
Differences in results in experiments in vivo have been attributed to variations in availability and distribution of alkaline phosphatase (Wills et al. 2007), cell lineage (Vellon et al. 2005), tirosine kinase activity, redox status, cell cycle stage, transcriptional regulation of apoptosis and DNA repair genes (Gloc et al. 2002).
Alkaline single cell gel electrophoresis Tice (1995) or “comet assay” was performed in leukocytes from normal mice pretreated with different doses of amifostine, so as to determine if the aminothiol in the therapeutic range was able to protect DNA against cisplatin in vivo. Superoxide dismutase (SOD) and catalase (CAT) activities as well as malonyldialdehide (MDA) were also measured in serum, in order to evaluate oxidant status regarding cisplatin and amifostine treatments.
MATERIALS AND METHODS
Animals
Mice were purchased from the Center for Production of Laboratory Animals (CENPALAB, Cuba). Sixty adult male from OF1 line weighing 25–30 g were placed 5 per box with filtered water and food (RatoninaR CENPALAB) supplied ad libitum. Conditions at room were: temperature, 26° C, humidity 50 % and 12 hours light / darkness cycles. All the animals were let in their cages for a week before the beginning of the experiments.
Experimental design
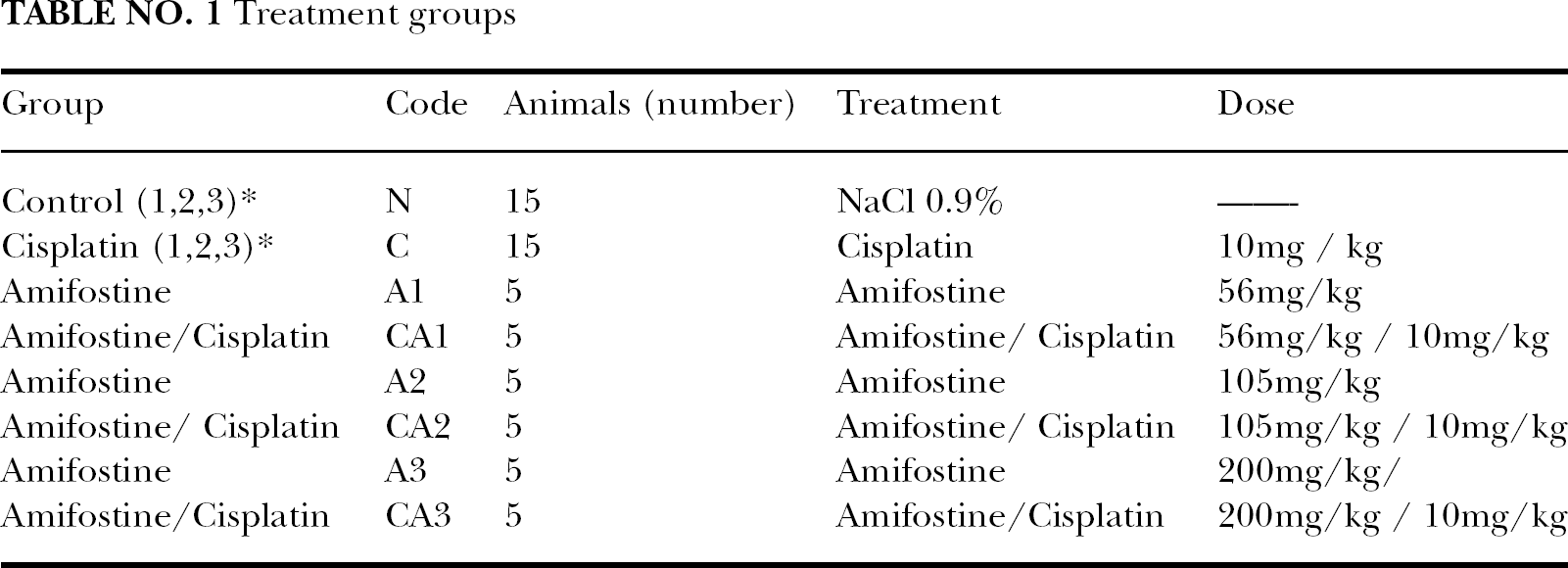
Animals were divided in four groups according to treatment: negative control (N), cisplatin (C), amifostine (A), cisplatin plus amifostine (CA). Amifostine (Ethyol) was assayed at three doses by ip injection near the therapeutic range. (Kouvaris et al. 2007) Table 1 Doses were 56, 105 and 200 (mg/kg). Cisplatin (LEMERY. Mèxico) was also given ip (10 mg/Kg) 15 minutes after amifostine. Animals were placed in their cages for 24 hours, and then were killed by cervical dislocation.
Treatment groups
Cells sampling
Blood samples were taken from. tail vein immediately before sacrifice. Samples were heparinized and peripheral blood leukocytes (PBL) for comet assay were isolated by routine density gradient centrifugation over Ficoll-Paque Plus (Pharmacia Biotech AB, Uppsala, Sweden). Contaminating red blood cells were lysed, and PBL were washed and counted. Freshly isolated PBL were kept between 2 to 4° C for 5 min up to the procedure began.
Cell viability
It was determined with the Trypan Blue exclusion method 10 min after sampling. Cell viability was found to be high enough for genotoxicity testing.
Comet assay
Alkaline single cell gel electrophoresis was performed according to Singh et al. 1988 technique with modifications. In brief, 10 000 PBL suspended in 10 μl of Ca 2+ and Mg 2+ free phosphate buffer saline, pH 7.4.(PBS) (Sigma) were mixed with 75 μl of 0.75 % low melting point agarose (BDH) dissolved in the same buffer at 37° C, and layered over a microscopy slide precoated with 100 μl of 1 % normal melting point agarose. Low melting point agarose was let harden at 4°C for 5 min and another LMP agarose layer was added. After 5 min at 4° C, the slides were immersed in alkaline lysis solution (2.5 M NaCl, 100 mM Na2EDTA, 10 mM tris, 1 % N-laurylsarcosinate, 10 % DMSO and 1 % Triton X-100, pH 10) for one hour at 4<° C. Slides were immersed in 4° C fresh alkaline electrophoresis buffer (0.3 M NaOH and 1mM Na2EDTA, pH 13.5) for 20 minutes to allow DNA in the nucleoid to unwind. Electrophoresis was done at 25 V (1V /cm) and 300 mA) for another 20 minutes. The slides were washed three times for 5 min. with 0.4 M tris-HCl pH 7.5 at 4°C, excess of humidity was removed, and slides were kept in a dust free box until stained with silver nitrate (Sigma) at 10 μg/ ml. (Reinhardt-Poulin et al. 2000). All the procedure was performed under dim light.
Analysis of slides
Scoring was performed by the same technician following a simple blind procedure. Microscope Accu-Scope 3004 was used in order to analyze 50 cells per animal (250 cells per dose). Classification of comets included three arbitrary damage levels according to the amount of DNA in the “comet” tail: level 1: low damage, 5–20 %; level 2: medium damage, 20–40%; level 3: high damage, 40–90 %. Cells in level 2 and 3 were considered damaged. “Comets” with more than 50% of material in the tail and no nuclei detectable was classified as “clouds” and not scored. Criteria were modified from Anderson et al. 1994.
Determination of thiobarbituric acid reactants (TBARs)
Lipid peroxidation was measured by the formation of MDA with the thiobarbituric acid method described by Yagi (1984); using MDA-bis-dimethyl acetal (Aldrich Chem., Milwaukee, Wi) as a standard. Briefly, 0.2 ml of 7 % SDS, 0.2 ml of 0.1 N HCl, 0.2 ml of 10 % phosphotungstic acid and 1 ml of 0.67 % thiobarbituric acid aqueous solution were added to the serum. The samples were immediately heated at 952 C for 60 min. After cooling, the dye was extracted with 5 ml of n-butylalcohol by shaking vigorously. The organic phase was separated by g for 10 min. Fluorescence intensity of the organic(centrifugation at 4500 x phase was measured at excitation of 515 nm and emission wavelength of 553 nm.
Superoxide dismutase activity measurement (SOD)
SOD activity was determined in serum by monitoring spectrophotometrically at 560 nm the rate of reduction of nitroblue tetrazolium (NBT) by O2-., using a hypoxanthine-xanthine oxidase system as the source of superoxide anion (O2-.)(Spitz and Oberley 1989).
Catalase activity measurement (CAT)
CAT level was measured by the method described by Aebi (1984). A volume of 0.1 ml of serum was added to a cuvette containing 1.9 ml of 50 mM phosphate buffer (pH 7.0). In order to start reaction 1.0 ml of freshly prepared 30 mM H2O2 was added. Extinction rate of H2O2 was measured spectrophotometrically. at 240 nm. Activity of CAT was expressed as μmol H2O2 metabolized/ mg protein/ min.
Statistical analysis
A one way ANOVA and Chi square test were used to determine statistically significant differences amongst different treatments or drug associations (Duez et al. 2003).
RESULTS
Cell viability was over 60 % in all treatments except that of cisplatin alone. Negative control: 79 %; Cisplatin: 40 %; Amifostine (200 mg/ kg.): 72 %; Amifostine (200 mg/ kg plus cisplatin 10 mg/ kg.): 69 %
Basal DNA damage in unexposed murine peripheral blood leukocytes yields 64.6% comets in level 1 i.e. undamaged cells, whereas in animals exposed to a cisplatin single dose of 10 mg/ kg a significant increase in DNA in comet tails over the baseline values (p< 0.001) accounts for 98.2 % cells in categories 2 and 3 but only 1.2 % in level 1 of DNA migration.
Amifostine treatment at the lower doses A1 (56 mg/ kg) and A2 (105mg/kg) yielded 37.6 % and 45.1 in levels 2 and 3 (Damaged cells) a result not significantly different from that of controls. The highest dose, A3 (200 mg/kg) resulted in and 60.5% damaged cells, an increase that was significant compared to negative control. (p< 0.05)
In the combined treatment (CA= cisplatin/ amifostine), proportion of damaged cells increased along with the amifostine dose: CA1 61.4 %, CA2 67.2 % and CA3 70.9 % respectively, but this increment was only significant respect to negative control in CA3.
It should be stressed that the amount of damaged cells in the animals given only amifostine was significantly smaller than that found in all treatment with amifostine plus cisplatin (chi square 967, 8 y p = 0). In fact all treatments (negative control, cisplatin, amifostine, amifostine plus cisplatin) differs significantly in distribution (chi square = 49, 69; p < 0.001). Table 2.
DNA damage distribution in each treatment group

250 cells were analyzed per dose
Significantly different from cisplatin p< 0.001
Significantly different from negative control p< 0.01
Significantly different from negative control p< 0.001
In order to evaluate the effect of treatments on lipoperoxidation, MDA levels were assayed in serum, as can be seen in Table 3. Lipoperoxidation increased significantly with cisplatin infusion; also amifostine treatment raised MDA levels over the baseline although not significantly over the control values, Amifostine, in the presence of cisplatin, produced no effect on MDA content in the lower doses (A1, A2). However, the highest doses resulted in a paradoxical induction of lipoperoxidation in serum.
Serum oxidative stress biomarkers according to treatment

Significant difference with negative control. p<0.001
Activity of antioxidant enzymes SOD and CAT were unchanged with the sole exception of CAT increase CA3 combined treatment (Amifostine 200 mg/ kg plus Cisplatin 10 mg/ kg).
DISCUSSION
WR1065, the active form of amifostine, has been recognized as a cyto-protector against ROS generating drugs (Koukourakis 2002); Church et al. 2004; Jatoi et al. 2004; Kouloulias et al. 2005; Majsterek et al. 2005). A reduction in DNA damage should be expected in cells from animals pretreated with amifostine and then exposed to a prooxidant like cisplatin, especially when the genetic damage is evaluated through a highly sensitive technique as the alkaline comet assay, Tice (1995).
Alkaline comet assay reveals single strand breaks, alkali labile or abasic sites in DNA (Tice et al. 2000). Extensive DNA damage is expressed as a greater migration of DNA in the comets tail when cells are electrophoresed. When PBL from a cisplatin-treated group were analyzed with this procedure, up to 92.8 % cells were found damaged, as a result of the oxygen free radicals generated by cisplatin driven ROS attack to DNA backbone or as a results of incisions made by Nucleotide Excision or Base Excision Repair during oxidative adduct removal, Sancar (1994).
Cisplatin is known as a ROS generator; Siomek et al. 2006 showed that this drug increased the level of 8-oxoguanosine and 8-oxodesoxiguanosine in urine from patients under chemotherapy. Several groups have also detected diverse damaging effects of cisplatin over DNA, like adduction, fragmentation and crosslinking (Ferrer et al. 2003; Goodisman et al. 2006; Nadin et al. 2006)
DNA was damaged in most of the cells exposed, so the cisplatin dose was high enough to be a meaningful challenge to murine PBL genetic material. Therefore, any significant reduction in the proportion of damaged cells, or a shift to lower levels in DNA migration, should be attributed to amifostine protective effect. Our results, reveals that amifostine afforded protection over peripheral blood lymphocytes DNA.
Basal damage, defined a low baseline of 64.6 % undamaged cells, remarkably 22.2 % of comets in level 2 The lower dose A1 (56 mg/ kg) resulted in almost a similar percentage of undamaged cells than in the control group. (62.4 % amifostine lower dose vs 64.6 % untreated cells). Whereas in A2 undamaged cells also prevailed.
Is well known that amifostine is a DNA protective agent against cisplatin. WR-1065 the active form of amifostine can scavenge superoxide anions and peroxyl radicals and hydroxyl radicals, extremely aggressive specie that reacts with high rate constant with several intracellular targets like DNA. Blocking hydroxyl radical attack is one of the most beneficial actions of amifostine and is expressed as a diminishing in single strand breaks and consequently in DNA migration in the comet assay.
In cells from amifostine pretreated mice and then exposed to cisplatin there was a significant reduction in damaged cells in the two lower doses (CA1, CA2) related to those of cisplatin-treated ones (p< 0.001).
Damage level distribution with all amifostine doses shifted to lower categories compared to cisplatin-treated cells.
Protection by amifostine against oxidative DNA breaking agents has been documented using in vitro models. Buschini et al. 2000, using the comet assay found that pretreatment with WR-2721 protects white blood cells against melphalan. Muller et al. 2004, reported that amifostine at a dose of 250–5000 μg/ml diminished DNA migration in the comet assay on human leukocytes exposed to X rays, but this effect was strongly dependent from exogenously added alkaline phosphatase. Blasiak et al. 2002 have reported that 14 mM amifostine protects peripheral mononuclear cells against idarrubicin, while Majsterek et al. 2005 confirmed that idarrubicin induced DNA damage decreased in normal human lymphocytes pretreated with amifostine. Conversely Sadowitz et al. 2002 found that amifostine at clinically achievable concentrations did not affect the rate of leukocytes DNA platination in vitro.
In one of the scarce in vivo studies, amifostine decreased micronuclei frequency in bone marrow polychromatic erythrocytes from ciclophos-phamide treated Swiss mice (Mazur and Czyzewska 1994). Bergström et al. 1999 found that amifostine lowered DNA adducts formation rate in normal brain and kidney cortex in cisplatin treated rats. There have been also negative results regarding amifostine antigenotoxicity in vivo. When Balb/c mice were injected with 6 mg/kg idarrubicin and, 250 mg/kg amifostine diminished cytogenetic damage in bone marrow cells DNA as expressed in less micronucleated cells, but when DNA damage was evaluated through comet assay, amifostine did not protected peripheral blood monocytes nor splenic cells, (Campos Nebel et al. 2002). This report is in agreement with our result with the highest aminothiol dose
In the present work, the increase in DNA migration at the highest dose (200 mg/kg) resembled the enhancement of bleomicyn clastogenic effect on human lymphocytes pretreated with amifostine (Littlefield and Hoffmann 1993). Also synergistic effects of amifostine and radiation, producing oxidative stress in normal cells have been documented (Brenner et al. 2003).
According to data in Table 3, treatment with amifostine alone was not able to induce significant increments in SOD, CAT or MDA, however, with the combined treatment; the highest dose of amifostine does not exert the expected protective action against cisplatin toxicity. Combined treatment at the highest amifostine dose (CA3), on the contrary, elicited a significant increase in lipoperoxidation and catalase activity over the negative control. CA3 treatment (200 mg/kg) does not protect DNA but seems to evoke a prooxidant behavior. There are antecedents in literature that 200 mg/kg is a toxic dose in certain animal models (Tabachnik Schor, 1987; Schor, 1988; Klutmann et al. 2000; Brenner et al. 2001). Those findings have supported different hypotheses regarding to amifostine metabolism that generates hydrogen peroxide; the inhibition of polyamine synthesis by its affinity to polyamine transporter; disturbances in thiol homeostasis with consecutive increase of susceptibility to ROS, also driven DNA damage. (Giannopoulou and Papadimitriou, 2003; Grdina et al. 2000).)
Our results are in agreement with reports of additive effect between amifostine and oxidants when is given at doses of 200 mg/kg or higher. Hidroxidopamine when applied in combined treatment with amifostine (200 mg/kg) to C57BL/6 mice have shown glutathione depletion, leading leukocyte toxicity. Biphosphonates linked radionuclides plus amifostine (200 mg/kg), also provoked leukotoxicity in rabbits in two different experimental series (Klutmann et al. 2000; Brenner et al. 2001)
Leukotoxicity have been explained as the result of “intrinsic mielo-toxicity” of amifostine. In this paper we present evidence that toxicity to leukocytes could be related to a genotoxic primary damage over DNA.
There are several papers indicating that amifostine is able to reduce the MDA content in cells treated with different oxidative stress inducer agents (Perret et al. 1994; Stankiewicz and Skrzydlewska 2003; Bolaman et al. 2005). However there are results indicating that amifostine exhibited scavenging activity against spontaneous lipoperoxidation but not against to what was induced by iron-ascorbate (Marzatico et al. 2000). An example of amifostine paradoxical association with oxidative stress is the enhancement in glutation peroxidase activity, interpreted as a response to ROS increase, following 375 mg/ day i.v amifostine in human cancer patients (Mantovani et al. 2003).
In the present work amifostine treatment did not increase significantly MDA over control values, nor in animals treated with cisplatin plus amifostine at 56 and 105 mg/kg. It is in agreement with the reported ability of WR1065 to limit lipid peroxidation (Stankiewicz et al. 2002) However MDA content increased significantly at the higher dose (CA3) result that can be linked to the higher genotoxicity exhibited at 200 mg/kg of amifostine alone or combined and can be explained as a threshold effect triggering some of the possible damage mechanisms mentioned above.
Regarding SOD activity, we did not show any significantly increased activity with amifostine, or even to cisplatin, our results are in agreement with reports on poor SOD inducibility or diminishing enzyme activity, perhaps due to oxidative damage to protein, in several oxidative stress models. From another line of evidence is known that Mn SOD mainly is inducible through transcriptional regulation by different stimuli and also amifostine is able to induce Mn SOD though NFk-β mediated mechanism but we did not measured that isoform. (Murley et al. 2004; Atalay et al. 2006)
Ability to alter redox status and induce changes in gene expression can explain differences in the cytoprotection provided by amifostine in diverse systems (Ortiz et al. 1999; Santini (2001). Altering p53 expression is one of the ways by which the aminothiol can alter redox status. Also upregulation in redox sensitive genes with NFk-β as transcription factor, unleashes a cascade of gene activation that could promote ROS production (Giannopoulou. and Papadimitriou, 2003; Lee et al. 2003; Murley et al. 2004; Khodarev et al. 2004; Jänicke et al. 2008).
We have not found reports on the use of the comet assay to evaluate amifostine protection against cisplatin in vivo; in our model amifostine protected leukocytes DNA at lower doses. Clinical Guidelines recommend doses in a range from 740 to 910 mg/m2. Our maximal dose (200 mg/kg) corresponds to 630 mg/m2, that is in the therapeutical range. The use of high amifostine doses in clinical practice should be considered cautiously It seems that 200 mg/kg is a threshold dose for toxic effects. Our results confirm the noxious effects of combined treatment at high doses and could help to explain previous reports of leuko and mielotoxicty as an the consequence of an increase in DNA primary damage.