
Abstract
Working under the assumption that hormesis is triggered by specific types of DNA damage, this report focuses on the types of damage which form the signature of ionizing radiation. The key attribute of the signature is the clustering of damage, arising from clusters of energy deposition such that more than one site within a 10 base pair segment of DNA has been chemically altered. A brief overview is given on what is currently believed to be the primary components of clustered damage produced by the direct effect. The overview draws primarily on studies that utilize electron paramagnetic resonance to measure free radical intermediates and gel electrophoresis to measure clustered damage in plasmid DNA. Based on this information, the threshold for a radiation induced biological response is calculated.
INTRODUCTION
Underlying any biological response to ionizing radiation there must be chemical changes. Because DNA is the critical cellular target (von Sonntag 2006), it is most likely that it is DNA damage that serves to trigger a response. More specifically, because clustered DNA damage is the unique attribute that makes ionizing radiation of particular importance in biology and medicine, the dose threshold for observing a biological response, and perhaps hormesis, should correlate with yields of clustered DNA damage. In this paper we give an overview of what is currently understood about the radiation physics and chemistry of DNA and use that information to estimate the threshold for a biological response.
The evidence continues to grow in support of the proposal by Ward (Ward 1981) and Goodhead (Goodhead 1990) that the main biological impact of ionizing radiation is that it creates lesions in DNA in the form of clusters. A lesion located near to others, i.e., in a cluster, is more prone to misrepair and is more likely to destabilize the genome (Shikazono, et al. 2006; Wallace, et al. 2004; Weinfeld, et al. 2001). Predicting biological response from first principles requires a quantitative understanding of the types of lesions formed and their spatial distribution. The degree to which lesions are clustered is a consequence of two factors, (i) the non-homogeneous deposition of energy that results in a track of initially altered sites, and (ii) the subsequent expansion of the track by diffusion of these altered sites. The indirect effect, a consequence of reactions between DNA and radicals generated in unbound water, requires diffusion of hydroxyl radicals from the water phase to the DNA target (see Figure 1). In contrast, the damage due to (what is loosely called) the direct effect, comes from precursors created by ionization of DNA and its solvent shell. The mean diffusion distance of these precursors is very short (Spalletta and Bernhard 1992; Yan, et al. 1992), < 1 nm (i.e., less than 3 base pairs) and consequently the damage has a high probability of being clustered. The direct effect, therefore, plays a major role in the fact that an imprint of the track is left on DNA (Nikjoo, et al. 2001).
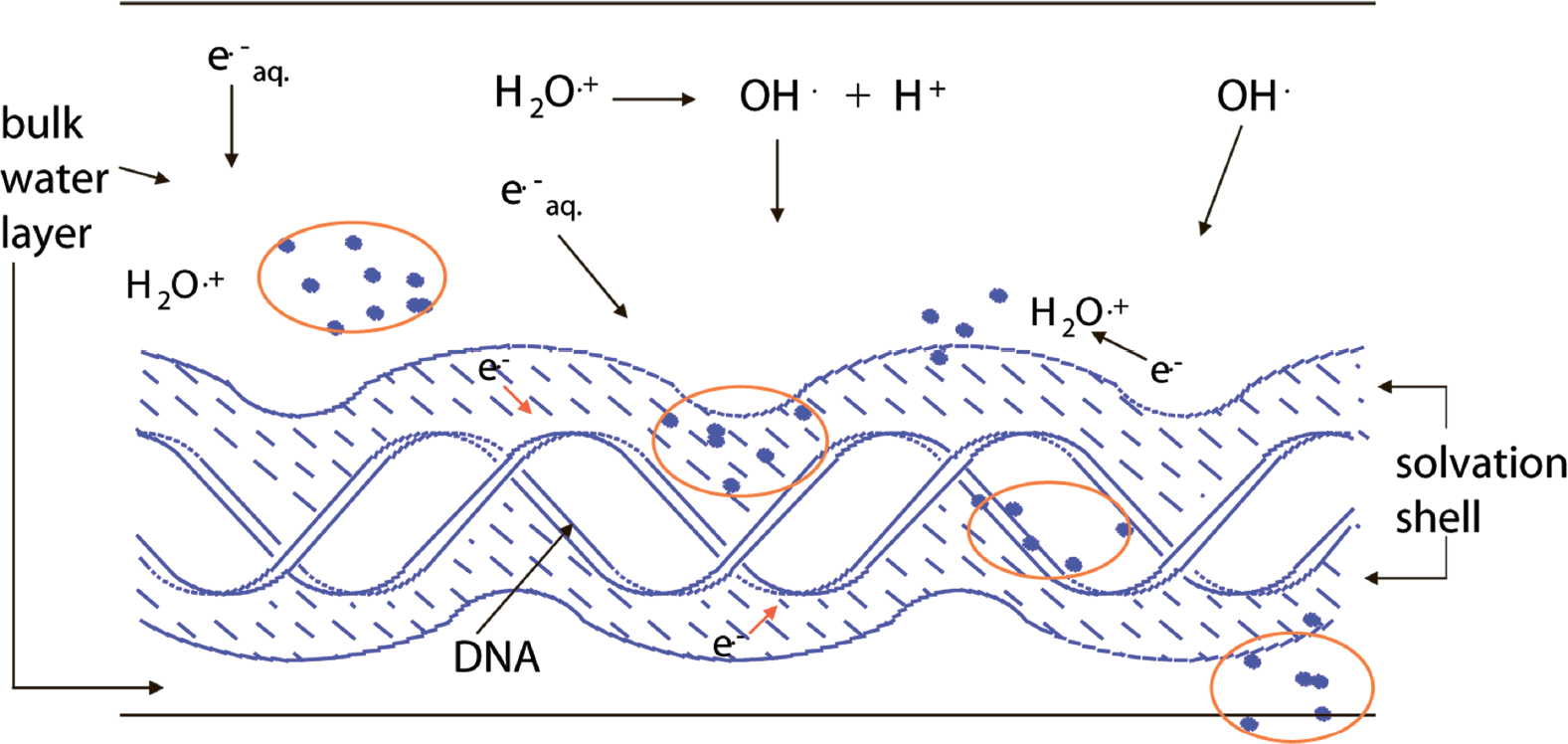
DNA in aqueous solution. Cartoon showing the intersection of an ionizing radiation track with DNA, the solvation shell of DNA, and the bulk water in the vicinity of DNA. The solid dots represent energy deposition events, i.e., ionization and excitation. Ionization of bulk water yields the water radical cation, H2O+·, and the aqueous electron, e− aq, which may react with DNA giving indirect-type damage. Ionization of DNA and its solvation shell results in direct-type damage. The DNA solvation shell consists of up to ∼20 to 22 waters/nucleotide.
The indirect effect of ionizing radiation was considered in the early radiobiology literature (von Sonntag 2006) to be more biologically relevant than the direct effect. Based on current knowledge, however, it is more likely that the direct effect is of comparable or even greater importance. Direct-type damage is formed via two routes. One is by direct ionization of the DNA and the other is by ionization of that portion of the solvent shell that is tightly bound to the DNA. Although the later is physically an indirect effect, by transferring holes and electrons created in the DNA solvation shell to the DNA, the initial DNA lesions are indistinguishable from those formed directly in the DNA. Damage formed via electron or hole transfer from the solvation shell has been termed, therefore, the “quasi-direct” effect (Becker and Sevilla 1993). The damage created by both routes is called collectively “direct-type” damage (Milano and Bernhard 1999), distinguishing it from “indirect-type” damage that is due to reactions with hydroxyl radicals and aqueous electrons. Proof and quantification of damage transfer to DNA from its solvation shell (Becker, et al. 1997; Debije, et al. 2000; Milano and Bernhard 1999) has proved quite important. Inclusion of the solvation shell effectively doubles the target mass resulting in direct-type damage. This doubling is used in the calculation reported in the final section of this paper. The fact that the solvation shell magnifies direct-type damage, and its importance in the formation of clustered damage, is not yet fully appreciated.
There are good reasons to believe that the composition of clusters is dominated by direct-type damage. It is often quoted that the direct effect accounts for between 30% and 50% of the damage in vivo. As reviewed by von Sonntag (von Sonntag 2006), it is difficult to design experiments capable of dividing the direct effect from the indirect effect; this is particularly true for in vivo measurements. In our view, one of the better estimates of the relative contribution of the direct-effect is that by Krisch, et al. (Krisch, et al. 1991). Using a well defined system consisting of SV40 viral DNA and varying the scavenger efficiency by over four orders of magnitude, they measured ssb and dsb with gel electrophoresis. Moreover, they compared the yields of ssb/dsb at −75°C (where the direct effect predominates) with those at 0–20 °C. They estimated that under physiological conditions ∼50% of the damage would be due to the direct effect. This percentage was based on only one type of damage, the deoxyribose damage that results in ssb and dsb. From our recent studies (Purkayastha, et al. 2007), less than 40% of the clustered lesions are of the type that would be revealed as dsb in the Krisch experiments. In addition, we have shown that damage at C1′ of the deoxyribose is not readily revealed as an ssb (Roginskaya, et al. 2005a; Roginskaya, et al. 2005b), and it would have most likely been missed in these experiments. If experiments were designed to take these two observations into account, the likely outcome would be that the lesions making up clusters are predominantly formed from direct-type damage. These estimates pertain to low LET radiation; at high LET, direct-type damage would account for an even larger fraction of the clustered damage (Becker, et al. 1996; Nikjoo, et al. 1998; Roots, et al. 1990; Yokoya, et al. 2003).
That direct-type damage accounts for a significant fraction of all of the damage to DNA in vivo should not be surprising. In eukaryotes, DNA is tightly packed within chromatin, conferring upon it mixed properties of the aqueous state and solid state. Measurements of the concentration of DNA in the nucleus over a wide range of eukaryotic cells, give an average of ∼100 mol H2O/mol nucleotide (Daban 2000). Given that: (i) 10% of this water is an integral part of the DNA target, (ii) some fraction (∼50%) is part of the protein solvation shell, and (iii) the very high protein concentration scavenges a large fraction of the HO•, one would anticipate that the direct effect accounts for about half of the DNA damage and more than half of the clustered damage.
REACTION PATHWAYS LEADING TO CLUSTER FORMATION BY THE DIRECT EFFECT
Clusters are formed when secondary electrons reach energies low enough that electrons created by further inelastic collisions undergo addition collisions in close proximity to one another. In the track structure field, these are called spurs and blobs (Magee and Chatterjee 1978). Once thermalized, the samples contain a non-homogeneous distribution of ionized sites. The track consists of trapped ions and electronically excited molecules. With respect to DNA damage, excited states appear to be negligible (Bernhard, et al. 1994). The trapped ions consist of one-electron-loss sites (free radical cations) and one-electron-gain sites (free radical anions). We refer to these as holes and electron-gain sites, respectively. These free radicals are inherently unstable, reacting quickly at room temperature to give stable diamagnetic damage which cannot be detected by electron paramagnetic resonance (EPR) spectroscopy. The challenges posed by the instability of free radical intermediates have been met by studying radicals trapped in solid state DNA at low temperatures using EPR.
There is without question more than one type of reaction pathway giving rise to direct-type damage. The pathway presented here is one that we believe is likely to account for the largest fraction of end-product. We call it the trappable-radical single-track pathway (Swarts, et al. 2007); other pathways are mentioned below. The trappable-radical single-track pathway is one in which the initial radicals, formed by either electron loss or electron gain, escape the spur of ionizations. These isolated radicals can be trapped, their structures can be analyzed by cryogenic EPR, and the thermally driven reactions can be studied by heating. Eventually, the radicals form stable end-product in which all electrons are paired. These diamagnetic end-products can no longer be detected by EPR; other analytical techniques must be applied.
Our mechanistic model assumes that half of the radicals trapped at 4 K are derived from sites of electron loss, holes, and the other half from sites of one-electron gain (Figure 2). Within a few picoseconds of the energy deposition, the system is thermalized, and the electrons are captured exclusively by the bases. The holes, meanwhile, are initially formed on the sugar-phosphate backbone, the DNA solvation shell, and the bases. Holes, initially formed in that portion of the solvation shell directly bound to DNA, transfer to the sugar and bases (Becker, et al. 1997; Debije, et al. 2000). The four predominant radical types are dRib• (deoxyribose radical), Gua+• (guanine radical cation), Cyt−• (cytosine radical anion), and Thy−• (thymine radical anion). Their relative concentrations are about 1:4:4:1, respectively (Bernhard 1989; Bernhard and Close 2003; Purkayastha, et al. 2006). The structures of the free radical intermediates, stable end products, and corresponding labels are given in Figure 3. The radicals, with the exception of Thy−•, are unstable at 4 K and are not observed by EPR. By undergoing dielectric relaxation, consisting primarily of proton transfer to the radical anions and proton transfer away from the radical cations, the radicals are stabilized and readily studied by EPR (Debije and Bernhard 2002).


Chemical structures and notation used in the text.
The thermally activated reactions stemming from dRib• depend on which deoxyribose carbon has undergone the net loss of a hydrogen atom. In Figure 4, the reaction pathway for the C1' centered radical, dRib (C1'-H)• is given as a specific example. One-electron oxidation of the radical followed by OH− addition releases free base and leaves behind an intact DNA backbone containing deoxyribonolactone (dRibonoLac). The deoxyribonolactone lesion is relatively stable at room temperature; but, upon warming, it yields 5-methylenefuranone and a cleaved strand (Roginskaya, et al. 2005a; Roginskaya, et al. 2005b).

The deoxyribose radical cation, dRib+•, deprotonates from any of the five deoxyribose carbons. Shown here is an example of the reactions stemming from C1' deprotonation.
One of the major pathways initiated by one-electron oxidation of guanine is shown in Figure 5. The guanine radical cation, G+•, undergoes OH− addition creating the neutral G (C8+OH)• radical (Cullis, et al. 1996). Subsequent one-electron oxidation gives the well known product of 8-oxoGua. The fate of one-electron reduced thymine, Thy−•, is shown in Figure 6. It protonates at C6 to give the neutral Thy(C6+H)• radical (Wang, et al. 1997; Wang and Sevilla 1994), which when one-electron reduced results in the formation of 5,6-dihydrothymine, DHThy. The type of reaction sequence produces 5,6-dihydrocytosine from cytosine (Debije, et al. 2002), but in this case hydrolysis leads to deamination and the final stable end product is 5,6-dihydrouracil, DHUra.

The reaction pathway leading to the formation of 8-oxo-guanine starting from the guanine radical cation, Gua+•.
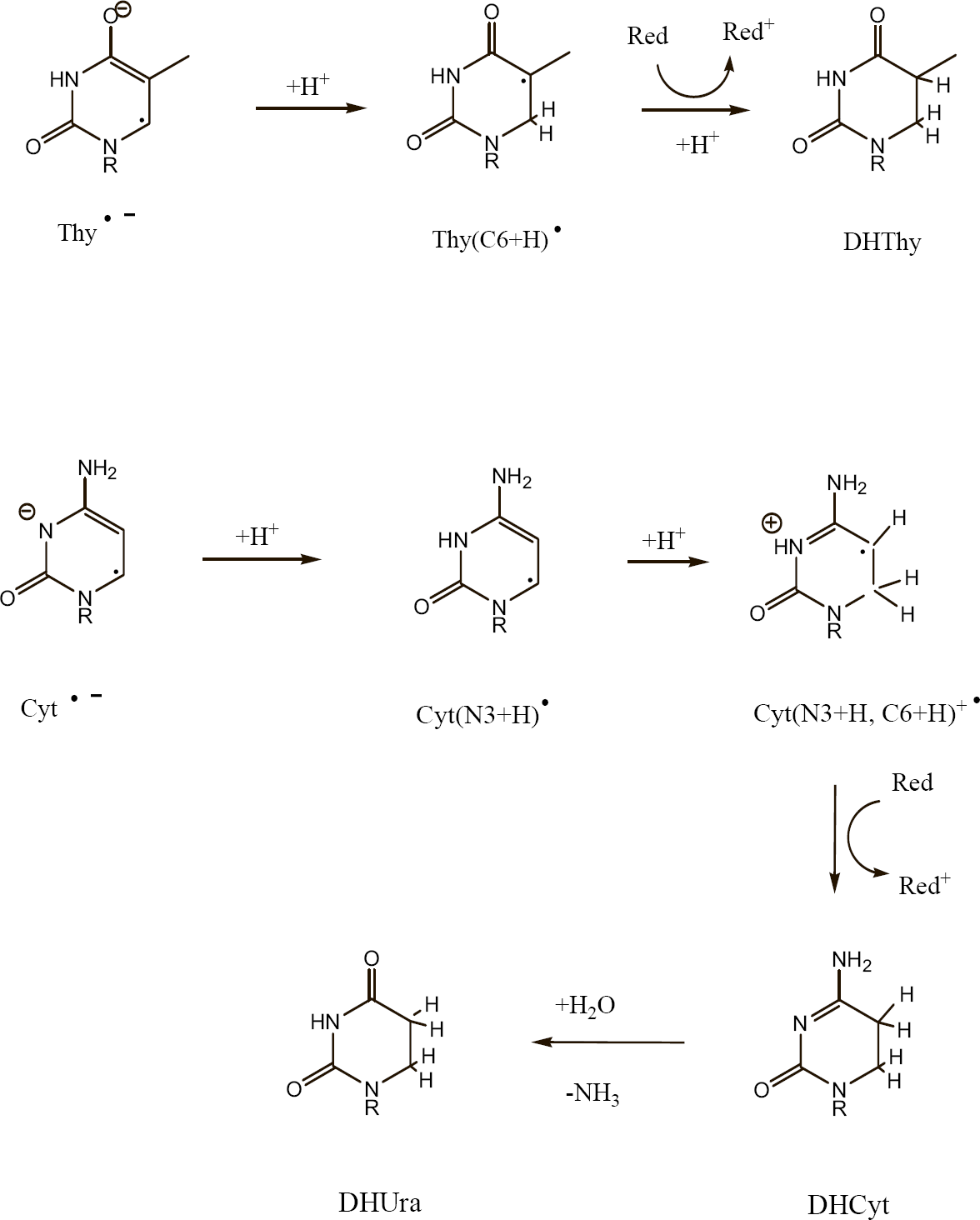
The reaction pathways leading to 5,6-dihydrothymine, DHThy, and 5,6-dihydrouracil, DHUra, starting from the respective one-electron reduced pyrimidines, Thy−• and Cyt−•.
By this model, four of the major lesions produced in DNA by the direct effect are explained. The four lesions are strand breaks, 8-oxoGua, diHUra, and diHThy (Swarts, et al. 2007).
The above model is not all inclusive. There are other mechanisms and other DNA products. The above pathway is initiated by inelastic collisions between the electron and DNA. Other pathways exist; an important set of reactions are those initiated by the elastic scattering of low energy electrons (LEE). LEE have been shown to undergo dissociative electron attachment (DEA) to DNA, producing single strand breaks (ssb) and double strand breaks (dsb), as well as fragmented bases (Huels, et al. 2003; Huels, et al. 1998; Li, et al. 2006). The yield of these end points, however, is not known. Ionization of the deoxyribose moiety can occur indirectly via the purines. It has been recently discovered that the guanine radical cation, when photoexcited by 520 nm light, undergoes hole transfer to the deoxyribose (Adhikary, et al. 2006; Adhikary, et al. 2005). The net result is a selective loss of hydrogen from C1', C3', and C5' of the deoxyribose. Furthermore, when radical cations are formed by direct ionization, they are initially left in an electronic excited state. Thus, it has been proposed that this reaction may occur in DNA due to the direct effect of ionizing radiation (Adhikary, et al. 2006). Swarts and co-workers observed a further 12 oxidation products in addition to those discussed above, for example 8-oxo-adenine (8-oxoAde), 5-OH-cytosine, and 5-OH-uracil (Swarts, et al. 1996). Molecular hydrogen has been measured as a function of hydration level (Falcone, et al. 2005). Interstrand crosslinks have been observed in halogenated DNA containing single stranded segments (Cai, et al. 2005).
PREDICTED THRESHOLD FOR A BIOLOGICAL RESPONSE
In recent work (Purkayastha, et al. 2007), we measured the yields of clustered lesions in DNA. The yield of DNA base damages due to the direct-type effects of ionizing radiation was compared with the yield of DNA trapped radicals previously measured in the same pUC18 plasmid across the hydration range of 2.5 to 22.2 mol water per mol nucleotide. Single strand breaks, ssb, and double strand breaks, dsb, were detected by agarose gel electrophoresis. Specific types of base lesions were converted into ssb and dsb using the base-excision repair enzymes, Endo III and FPG. The yields of dsb were consistent with changes in free radical trapping as a function of hydration. The composition of three types of clusters and the yields of each cluster type was determined. The yield of dsb due to deoxyribose damage on opposing strands was 3.5 ± 0.5 nmol/J for fully hydrated pUC18. For an oxidized purine or an abasic site on one strand and an oxidized purine, an abasic site, or a deoxyribose damage on the opposite strand, the yield was 3.2 ± 0.9 nmol/J. For a reduced pyrimidine or an abasic site on one strand and a reduced pyrimidine, an abasic site, or a deoxyribose damage on the opposite strand, the yield was >2.3 ± 0.9 nmol/J, where the inequality reflects the possibility that reductive damage is underreported in the protocol employed. Less than 40% of the clustered lesions are due to opposing deoxyribose damage, the damage that would be scored as prompt dsb. The yield for these three types of clusters is ∼10 nmol/J; this yield is employed in the following calculation.
The 46 human chromosomes are comprised of 7.8×109 bp, giving a molecular weight of 5.2×1012 Da assuming one Na+ per nucleotide. Because the hydration shell transfers direct damage into the DNA (see above) there are ∼22 waters per nucleotide that are part of the target mass. With respect to direct damage, the target mass is, therefore, ∼11×1012 Da. Using 10 nmol/J and a dose of 1 mGy, the experimentally obtained yield (for what are presumed to be three of the major types of clusters produced by the direct effect) is ∼ 1 cluster per 10 genomes. If damage to 1% of the cells is required to detect a biological response, then a threshold dose of ∼0.1 mGy is predicted. This estimate is based on clusters formed solely by the direct effect. If we assume that inclusion of indirect DNA damage doubles the yield of clusters, then the predicted threshold is ∼0.05 mGy.
In concluding, we compare these predicted levels of clustered damage with measured damage and damage responses in vivo. DSB were measured in human fibroblasts using immunofluorescence to detect γ-H2AX foci at very low doses of 90 kV x-rays (Rothkamm and Löbrich 2003). They found that a 1 mGy exposure resulted in 1 DSB per ∼30 cells. If we assume that DSB account for 40% of the clustered damage, as per the results described above, this corresponds to 1 clustered lesion per ∼ 10 cells; the same as predicted for cluster formation by the direct effect alone.
The yield of chromosomal aberrations is of interest because they are detected subsequent to substantial biochemical processing. In primary human fibroblasts exposed to 1 Gy of γ-rays, one aberration is detected per 20 cells (Cornforth, et al. 2002). It seems rather certain, therefore, that by the time chromosomal aberrations can be scored, the genome has sustained a level of clustered damage that is about three orders of magnitude larger than the number of aberrations. Better insight into the correspondence between clustered damage and biological endpoints, should be possible by finding models that are more sensitive to radiation exposure.
An example of a highly sensitive model is recombination mutagenesis in pKZ1 transgenic mice (Matsuoka, et al. 1991). Hooker, et al. used this model to study gene inversion in the spleen at very low doses of 250 kV X-rays administered to pKZ1 mice (Hooker, et al. 2004). They found a statistically significant gene inversion response of ∼2×10−4 per cell at 5 μGy while the response at 1 μGy was not significant. Based on our calculated threshold and given that the size of mouse genome is ∼86% that of the human genome, at 5 μGy the frequency of clustered damage is predicted to be ∼5×10−4 per cell. The prediction, thereby, is that one gene inversion is produced for every ∼2 cells that have sustained clustered damage. This implies that induction of recombinatorial repair is exceptionally sensitive to the presence of clustered damage or, as mentioned by Hooker, et al., that a bystander effect (Mothersill and Seymour 2001) may amplify the response.
Footnotes
ACKNOWLEDGMENT
This study was supported by PHS grants 2-R01-CA32546 (to WAB), and 2-R01-CA46295 (to JRM), awarded by the National Cancer Institute, DHHS.