
Abstract
Microwave (MW) fixation has been suggested as a method to rapidly immobilize cellular dynamics for fine structural studies in the electron microscope. To show its suitability for studies on cell monolayers, one has to apply MW fixation systematically in correlation with samples on the light microscopy level. Examples for MW fixation of cell monolayers, however, are still rare. MW-accelerated fixation for relatively long periods of time (1–2 min) has been reported without showing its suitability at the fine structural level. Here, we provide a rapid MW fixation protocol for cell monolayers on a subminute time scale. The impact of the MW-accelerated glutaraldehyde fixation on temperature-sensitive cytoskeletal components such as microtubules was evaluated. For testing the effectiveness of MW-assisted primary fixation, saponin treatment of the monolayers was included. Simultaneous MW-accelerated fixation and extraction by saponin was necessary to achieve a gradual improvement in visualization of cytoskeletal aspects in association with cell junctions, mitochondria, and centrioles. To establish a valuable routine program for fine structural studies of resin-embedded cell models on substrata, a protocol combining MW fixation with automatic processing in a tissue processor is provided.
Keywords
R
Because high-quality MW fixation is limited to a few micrometers at the periphery of bulky tissues (Wild et al. 1989), one can expect that its application to substrata-grown cell monolayers is favorable for structural conservation of such samples. Unfortunately, the MW fixation protocols for cell monolayers developed thus far (Kok and Boon 1992; McDonald 2002) have not been used to show their potential for visualization and preservation of cellular fine structures. Here, we present a novel and straightforward protocol for rapid MW-accelerated glutaraldehyde (GA) fixation of cell monolayers within a subminute time scale and show its suitability for studies at the level of cellular fine structure. Similar to the methodical developments for hippocampal tissue slices by Jensen and Harris (1989), we adjusted the MW-accelerated fixation of cell monolayers to temperatures that seem not to damage microtubules (MTs). As a consequence, we were able to show structurally intact MT-associated cell structures such as MT-tethering and adherence junctions (Ligon et al. 2001; Reipert 2007), mitochondria, Golgi complexes, and centromeres. In combination with MW exposure, we raised saponin extraction to a level that increased visibility of the cytofilaments and membranes. To offer a practicable solution for routine processing of cell monolayers, we tested MW fixation in combination with further processing of the samples in an automatic tissue processor.
Materials and Methods
Tissue Culture
PtK2 rat kangaroo kidney epithelial cells were grown in DMEM supplemented with 10% heat-inactivated fetal calf serum at 37C and 8% CO2. They were plated either on glass coverslips, 12 mm in diameter, or on Aclar plastics (EMS; Science Services, Munich, Germany) of the same size. The substrata were cleaned by HCl treatment for 2 hr and subsequent washing in distilled water and 70% ethanol. Before use, they were incubated in culture medium at 37C overnight. Cells were grown to 90% confluency and split 1:3 by use of trypsin/EDTA every third day. Prolonged culture of PtK2 at a state of confluency was avoided, because this would alter cellular morphology as indicated by a smaller size ratio between nucleus and cytoplasm.
Standard Chemical Fixation
PtK2 cells on Aclar or glass coverslips were routinely fixed by immersion in 3% GA in 0.1 M sodium cacodylate buffer, pH 7.3, for 30 min at room temperature (Svitkina and Borisy 1998). Fixation was followed by washing three times in sodium cacodylate buffer for 10 min each. For comparison with fixation parameters under MW exposure, fixation was also performed under conditions regarded as suboptimal (0.5% GA in sodium cacodylate buffer for both 30 min and 15 sec).
Calibration of the MW Oven for MW-accelerated Fixation
For MW-accelerated fixation, a pulsed MW oven (EMS-820; EMS, Hatfield, PA) with a pulse length of 3 sec and a magnetron power of 1000 W was used. MW exposure at a frequency of 2450 Hz followed the guidelines for a standardized application given by Login et al. (1998):
Since the built-in table for sample rotation was not used, it was covered by an oven tray. To ensure defined positioning of the sample and of the customized water load for protection of the magnetron, the oven tray was overlaid by a foil with an alpha numeric grid drawn on it.
Before every use, the magnetron was warmed up. One liter of water, placed in a plastic beaker in the center of the MW oven, was exposed for 2 min at 100% output power, and the resulting rise in temperature of the mixed water load was measured immediately afterward. An initial drop in output power by ∼20% was noticed just after first-time use of the MW oven. Importantly, the MW output power remained stable at ∼800 W for any of the subsequent experiments. This and any other temperature measurements were performed with a Testo 735-2 instrument (Testo; Vienna, Austria) equipped with an immersion probe TC type K.
To protect the magnetron during the fixation experiments, a customized water load of 200 ml distilled water in a 400-ml plastic beaker was placed at a marked position in one corner of the MW oven.
For positioning of the sample during fixation, a central “hot spot” of the lateral MW field distribution was identified. It was found by measuring the lateral variation in samples warming up under standardized conditions (50% MW output power, 15-sec MW exposure of 3 ml distilled water in a 25-ml beaker). The characteristics of the MW distribution were also studied by continuous exposure of a self-made neon bulb array (Login et al. 1998) at 100% MW output power. To create such an array on the floor of the oven, the bulbs were mounted at 2-mm intervals. They were briefly exposed to MWs (<1 min) to avoid heat damage. Bulbs located in “hot spots” lit up during MW exposure with various intensities. The remaining bulbs indicated regions of low MW intensity, ∼4 cm in diameter, by not lighting at all.
To keep the conditions for MW absorption constant, a 25-ml glass beaker filled with 3 ml fixative was used as a standard throughout the experiments.
To find relevant conditions for MW fixation, we adjusted MW output power and exposure time according to the warming up of the solution (3 ml 0.1 M sodium cacodylate buffer, pH 7.3). Temperatures too high had to be avoided to prevent heat damage to cell structures during MW fixation (Leong and Sormunen 1998). Initially, we tested whether a single MW pulse of 3 sec (programmed time, 5 sec) would be sufficient to raise the temperature significantly. Even at 100% MW output power, this was not the case. Surprisingly, two pulses (programmed time, 10 sec) at maximum power led to excessive warming >50C, perhaps indicating a technical problem in initiating warm-up of the magnetron for the very first magnetron cycle. Given the specificity of the EMS-820 MW oven, either two pulses (programmed time, 10 sec) at 60% output power, or three pulses (programmed time, 15 sec) at 50% output power fulfilled the condition for a moderate buffer warm-up from room temperature by 10 ± 2C and 15 ± 2C, respectively. As a consequence, the temperatures of the fixative measured (33 ± 1C and 37 ± 2C, respectively) remained <40C. Addition of 3% GA to the buffer solution did not alter MW absorption significantly.
MW Fixation
Under the standardized conditions described above, the following GA concentrations were tested: 0.05%, 0.5%, 1%, 3%, and 6% in 0.1 M sodium cacodylate buffer, pH 7.3. For comparison, a combination of 0.5% GA with 2.5% and 4% paraformaldehyde in cacodylate buffer was also studied. Table 1 summarizes the standardized conditions and parameters for MW fixation that provided the best results.
For MW-accelerated fixation, cells on Aclar or glass coverslips were transferred from the culture dishes into the glass beaker filled with 3 ml fixative at room temperature. The time span from releasing the coverslips into the fixative up to the start of MW exposure was ∼2 sec. For assessment of the primary fixation effect by GA cross-linking, the fixative was replaced by 0.1 M sodium cacodylate, pH 7.3, immediately after MW exposure. Samples were kept in the buffer (for at least 30 min) or they underwent saponin treatment before further processing. Although immediate removal of the sample from the fixative was a precondition for studies of the effect of MW exposure, for routine application, samples can remain in the fixative for longer time.
Saponin Extraction
For combined MW fixation and extraction of biological material, 0.5% saponin from Quillaja bark (Sigma Aldrich; Vienna, Austria) was added to the fixative (0.5% glutaraldehyde in 0.1 M sodium cacodylate buffer). MW exposure was performed for 15 sec (programmed time) at 50% MW output power under standardized conditions stated above. Immediately after MW exposure, the saponin/GA solution was replaced by sodium cacodylate buffer. After three washes, for 5 min each, the samples remained in cacodylate buffer until postfixation and embedding.
Standardized conditions for MW fixation

To test whether there is an effect of saponin on MW-fixed cell monolayers, 0.5% saponin in 0.1 M sodium cacodylate buffer was applied to samples fixed with GA at low concentrations (0.05%, 0.5%). Saponin treatment was performed either short term (20 min) or long term (18 h) at room temperature by agitation in a LEICA tissue processor (LEICA Microsystems; Vienna, Austria). Alternatively, saponin extraction was done by MW exposure one time at 15 sec and two times at 15 sec (programmed time) at 50% output power under the standardized conditions stated above. For immediate repetition of MW exposure, samples were transferred into fresh saponin solution at room temperature. Saponin treatment was followed by three buffer washes for 5 min each before postfixation with 3% GA in 0.1 M sodium cacodylate buffer, osmification, and embedding.
Postfixation and Embedding in Epoxy Resin for Comparative Studies
Subsequent processing steps of postfixation, dehydration, and epoxy resin embedding were applied to standard-fixed and MW-fixed monolayers in an automatic tissue processor (LEICA Microsystems). Four baskets (large size, three divisions) of the tissue processor were arranged in pairs of two at the stem assembly to provide space for the uptake of six coverslips in vertical position with the cells facing toward the stem (Figure 1). Postfixation with 0.5% OsO4, washing three times in 0.1 M sodium cacodylate, pH 7.3, for 10 min each, dehydration in an ethanol series (30%, 50%, 70%, 95%, and two times 100% ethanol for 10 min each), mediation of the resin infiltration by propylene oxide (two times for 10 min), and sample infiltration in mixtures of propylene oxide and Agar 100 (Agar Scientific; Cambridge, UK), were performed under permanent moderate agitation. After infiltration in the following solvent:resin mixtures, 1:3 for 15 min, 1:2 for 15 min, and 2:3 for 45 min, the coverslips were taken out of the baskets and placed on glass slides with the cell layers facing up. After infiltration of the cell layer in droplets of pure resin for 2 hr, an Eppendorf tube with its bottom and lid cut off was placed above the sample. After an initial heat polymerization at 60C for 3 hr, the Eppendorf tube was filled with resin, and polymerization was continued overnight. Separation of the hardened resin block from glass coverslips was done by immersion in liquid nitrogen and subsequent warming. Aclar, in contrast, could be lifted easily from the resin by applying minimal mechanical force (McDonald 2002). Every cylindrical resin block was cut into quarters perpendicularly to its abutting face. As a result, the fixation quality could be evaluated at four different locations of the same coverslip.

Automatic processing of coverslips in a tissue processor. (
Based on the technical details described above, the following experimental schedule was used: (a) MW primary fixation under variable conditions to limit warming up of the fixative by MWs to a maximum 40C (test of the MW oven settings and of the fixative concentration), (b) standard tests to challenge the stability of GA cross-linking by extraction (immediate replacement of the fixative by buffer solution and saponin), (c) standard epoxy resin embedding, sectioning, and contrasting, and (d) comparative morphological and fine structural assessment in the electron microscope.
For (d), three MW-fixed monolayers (up to 12 resin-embedded samples) were sufficient to spot major problems caused by suboptimal fixation parameters. The basic protocols for MW fixation with or without addition of saponin to the fixative, however, were tested intensely for 24 monolayers each. Thin sections were cut with an ultramicrotome Ultracut S (LEICA Microsystems), mounted on copper grids, counterstained with uranyl acetate and lead citrate, and examined at 80 kV in a JEM-1210 electron microscope (JEOL; Tokyo, Japan). Images were acquired using a digital camera (Morada; Soft Imaging System GmbH, Münster, Germany) connected to the wide-angle port of the transmission electron microscope and analySIS FIVE software (Soft Imaging System).
Results
We tested MW-accelerated GA fixation of cell monolayers at a subminute time scale. Central questions in establishing the protocol were (a) whether cellular morphology and fine structure could be preserved despite significantly shortened fixation time and the possibility of irradiation damage, (b) whether the results produced by MW fixation were reproducible, and (c) to what extent the visibility of cytoskeletal aspect could be increased.
Criteria for evaluation were overall cell morphology, the appearance of cytofilaments and membrane-bound organelles, and the distribution of ribosomes. Because we aimed at the visualization of MTs in association with organelles and cell junctions, we compared our result by MW fixation with conventional chemical fixation under conditions known to be favorable to preserve cytofilaments (Svitkina and Borisy 1998).
Establishing Working Parameters for MW-accelerated GA Cross-linkage
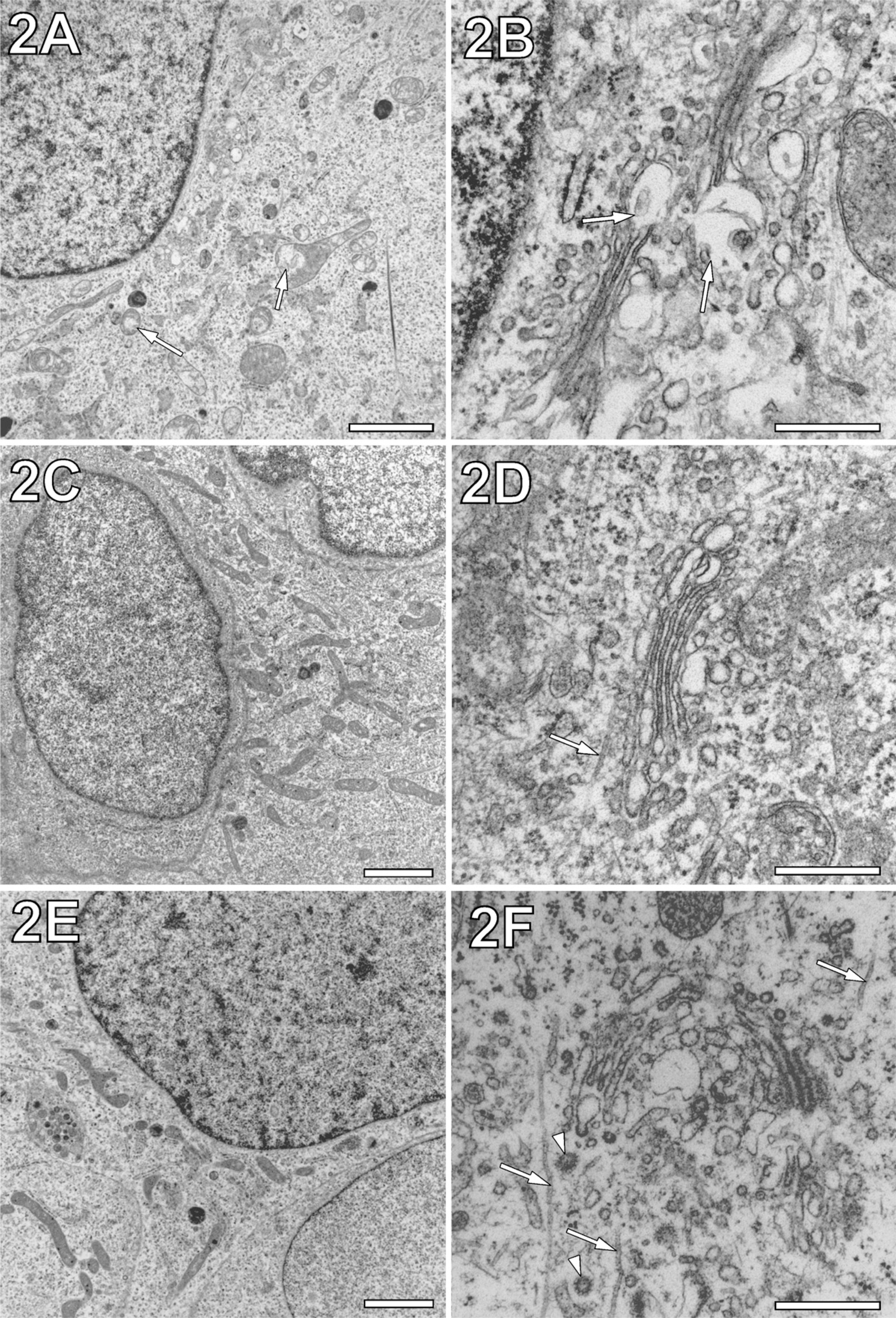
We aimed at the most rapid MW fixation schedule possible. Initially, we tested exposure of the sample in 3% GA fixative with just a single pulse of 3 sec (programmed time, 5 sec) at maximum power. Despite minimal warming up of the fixative by 2C, this resulted in severe damage to the mitochondria and significant loss of ribosomes. To succeed with cellular preservation in GA, either two or three MW pulses were needed. To avoid heat damage to cells in the given volume of fixative, the output power had to be reduced to 50% and 60%, respectively. Under conditions of a limited warming up to 10–15C, the overall cell morphology was always well maintained compared with conventionally fixed samples (compare Figures 2A and 2C). Notably, our MW-fixed samples did not show swelling of the rough endoplasmic reticulum (rER) or holes in the nuclei, which have been reported previously as a result of severe deterioration by MWs (Kok and Boon 1992).
Comparison of conventionally chemically fixed and microwave (MW)-fixed PtK2 cells. (
Besides the technical parameters of the MW oven, we studied the role of the fixative in MW fixation of the cell monolayers. First we tested whether MW irradiation of cell monolayers in a buffered solution without fixative would be sufficient to achieve fixation, as suggested by Argall and Armati (1990). MW exposure of 15 sec (programmed time) at 50% power and OsO4 postfixation after 10-min immersion of the sample in sodium cacodylate buffer could not prevent heavy damage to cells, indicated by blistered organelles, electron lucent cytoplasm, and barely recognizable remains of cytofilaments and cell junctions. Therefore, we concluded that cross-linkage by fixatives is essential to preserve cell monolayers by MW fixation.
Because previous applications of GA for MW fixation varied in a wide range of concentrations (0.05% and 6%), we wanted to know which GA concentration would best suit preserving the cell monolayers (Jensen and Harris 1989; Leong and Sormunen 1998). MW fixation with 0.05% GA, despite low fixative concentration, led to remarkably well-maintained overall cell morphology, whereas at the subcellular level, a large number of swollen and damaged mitochondria were apparent (results not shown). Hence, we considered such a low GA concentration insufficient to preserve cell monolayers adequately.
Compared with conventional chemical fixation over minutes, rapid MW fixation with 0.5% and 3% GA both resulted in excellent overall cell morphology throughout the samples (compare Figure 2A with 2C and 2E). However, similarly short conventional fixation with GA for 15 sec at room temperature led to heavy mitochondrial damage and loss of ribosomes. Therefore, to avoid unacceptable damage in conventional GA fixation, a fixation time of 30 min was needed (Figure 2A). Even under this condition, samples contained a higher number of blistered mitochondria than cells rapidly MW fixed with 0.5% and 3% GA (Figures 2C and 2E). As a result, we regarded the range of GA concentration between 0.5% and 3% as promising for the more intense fine structural evaluation of the impact of MW-accelerated cross-linkage (see next section).
Because a concentration of 6% GA has been reported to preserve MTs in hippocampal slices (Jensen and Harris 1989), we tested whether such a high concentration would be suitable for cell monolayers. We found that MW fixation of PtK2 cells with 6% GA for 15 sec led to suboptimal overall morphology and a decrease in visibility of cytoplasmic structures. In particular, ribosomes were no longer apparent (data not shown). These results are in agreement with previous studies showing inhibition in the formation of rapid cross-links by too high concentrations of GA in conventional chemical fixation (Griffiths 1993).
Rapid MW Fixation Preserves Membrane-bound Organelles and Cytoskeletal Structures
To discriminate between conventional chemical fixation and MW fixation of cell monolayers, a comparison was performed at the fine structural level. Only at this level did we find that MW exposure with three pulses (programmed time, 15 sec) was necessary to ensure reproducibility for membrane preservation of organelles. Although high-quality preservation of membranes could also be achieved by MW exposure with two pulses (programmed time, 10 sec), in this case, reproducibility in quality was diminished to an estimated 70%.
Preservation of cell nuclei by rapid MW fixation either with 3% GA or 0.5% GA was indistinguishable from that by conventional chemical fixation with 3% GA. Nuclear membranes were free of membrane blistering, and if cut at their periphery, they showed the lateral distribution of numerous nuclear pores (data not shown). In contrast to the nuclear membranes, the membranes of Golgi complexes in conventionally GA-fixed cells appeared to be more prone to excessive blistering (Figure 2B, arrows). By MW fixation with 3% GA and 0.5% GA, this excessive membrane blistering of the Golgi complex was avoided (Figures 2D and 2F). Notably, even after MW fixation with the lower concentration of GA, the coating of vesicles at the Golgi trans-face was preserved (Figure 2F, arrowheads).
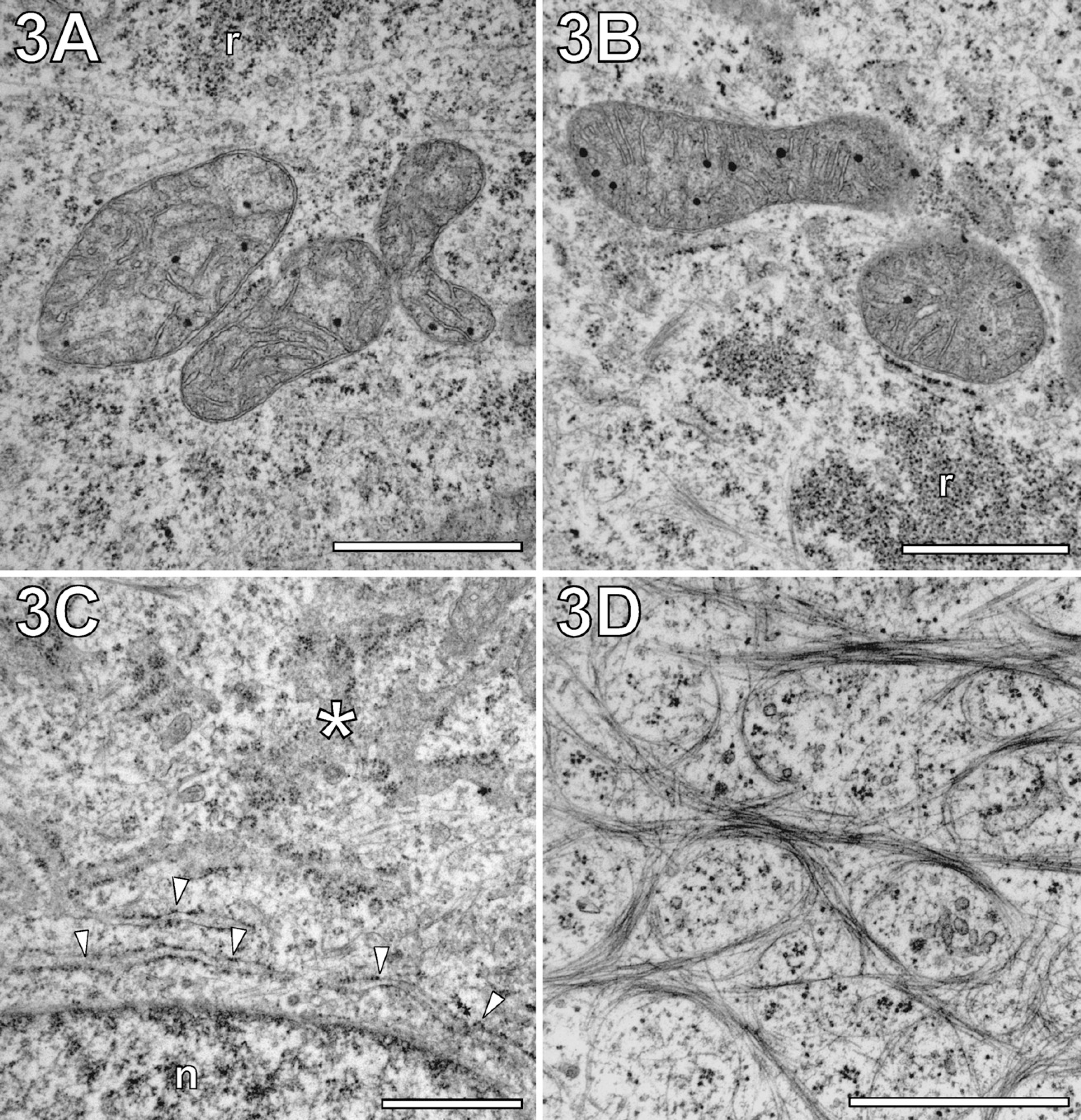
Although membrane preservation of mitochondria could be found already improved by MW fixation (see previous section), we also evaluated mitochondria at the fine structural level. Similar to conventional chemical fixation, MW-fixed mitochondrial profiles and their internal organization varied in a wide range. This result indicated a balanced cellular distribution of mitochondria in states of fission and fusion (for review, see Chan 2006). Comparison of rapid MW fixation with different concentrations of fixative (3% and 0.5% GA) provided similar fine structural preservation of cristae, matrix, and matrix granules (compare Figures 3A and 3B). Notably, the mitochondrial profiles shown in Figure 3 are devoid of membrane blistering. However, local blistering of membranes or partial vacuolization of tubular mitochondria could not be completely prevented by MW fixation. This might be related to either biological mitochondrial degradation or MW exposure-unrelated preparation artifacts.
Preservation of both membranous and filamentous aspects in PtK2 cells by MW-accelerated GA fixation. (
Regardless of the fixative concentration applied, the rER showed no signs of MW-induced deterioration. Figure 3C shows rER rapidly MW fixed with 0.5% GA in both confirmations, either organized as sheets with tubular extensions in parallel to the plane of section or cross-sectioned (Puhka et al. 2007). Moreover, numerous proximal arrangements of rER with mitochondria could be observed, which could possibly be functionally related to Ca2+ homeostasis (Pizzo and Pozzan 2007).
Preservation of organelles associated with MTs by MW-fixation/extraction. Cells were MW-fixed in cacodylate buffer containing 0.5% GA and 0.5% saponin. (
Regardless of whether conventionally fixed or MW fixed, GA treatment and subsequent embedding in epoxy resin made it difficult to visualize cytofilaments. Besides cortical actin filaments (see next section), only bundled intermediate filament (IF) networks contrasted against a rather electron-lucent cytoplasm (Figure 3D). This network closely resembled keratin IFs in keratinocytes and therefore probably represented the keratin-type filaments of PtK2 cells (Klymkowsky 1982; Osmanagic-Myers et al. 2006). MTs could only be spotted sporadically throughout the cytoplasm. Most frequently, they were found in association with organelles, such as the Golgi complex (Figures 2C and 2F, arrows). Testing the effect of Karnovsky-like combination of GA (0.5%) with paraformaldehyde (2% and 4%) for MW fixation, we found a strong dependence of MT contrasting on the composition of the fixative. The addition of paraformaldehyde led to negatively stained MTs, which contrasted against a rather electron-dense cytoplasm. At the same time, mitochondrial cristae showed an unusual irregular arrangement, which differed significantly from their appearance in high-pressure frozen PtK2 cells (Reipert et al. 2004). Because of these limitations, we decided to apply extraction by saponin for improved visualization of MTs.
Improved preservation of centrosomal MT asters by MW fixation/extraction. (
Improved Visualization of Organelles and Junctions in Association With MTs
Initially, we addressed the question of whether conventional or MW-enhanced extraction by saponin could be extensive enough to expose MTs and other cytoskeletal filaments in conjunction with organelles. For this purpose, we applied 0.5% saponin immediately after MW fixation at a low GA concentration (0.05% and 0.5%). Unfortunately, no whole mount–like structures could be obtained by this approach. Instead, for cells MW fixed with 0.05% GA, excessive damage to mitochondria became apparent, whereas the visualization of the cytoskeleton was barely improved. In samples MW fixed with 0.5% GA, improved preservation of mitochondria could be observed, whereas the action of saponin on the cytoplasm was hampered. Even a repetition of the MW-enhanced saponin extraction (see the Materials and Methods section) could not expose the cytoskeleton.
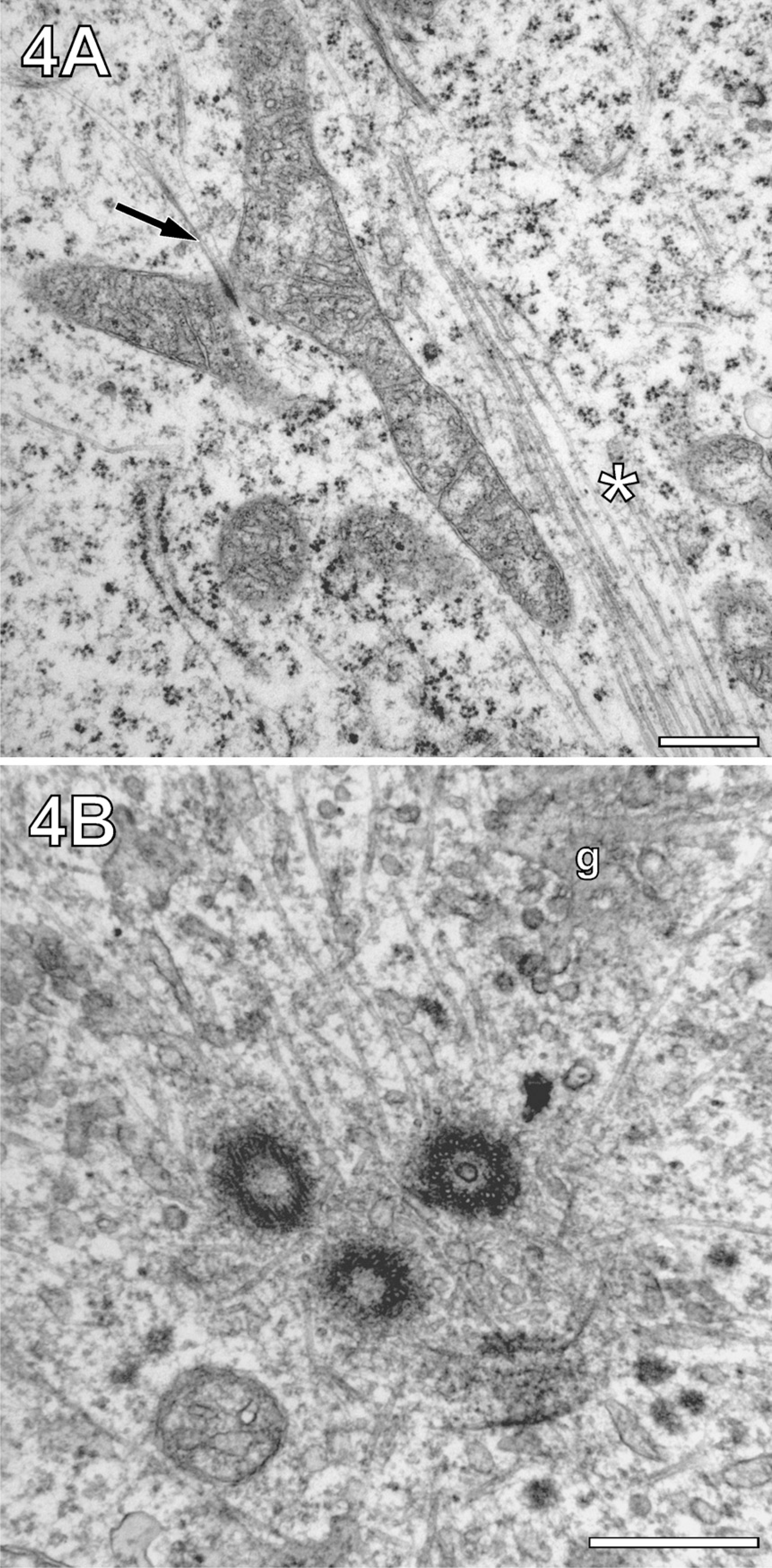
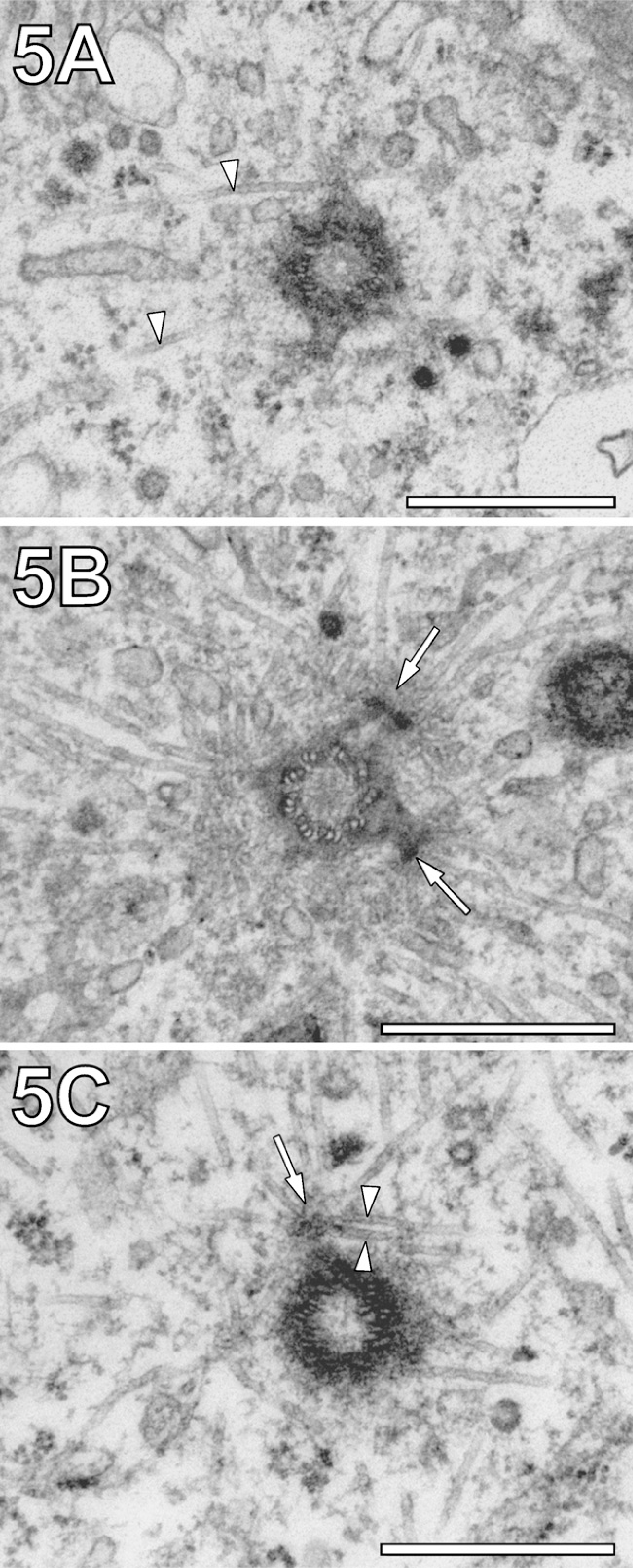
Because sequential saponin treatment after MW fixation failed, we exposed cell monolayers to MWs while they were in a mixture of 0.5% GA fixative and 0.5% saponin. This opened the possibility of visualizing MTs over larger distances (>1 μm) within a gradually extracted cytoplasm. Figure 4A shows the suitability of this method by showing an elongated mitochondrion in association with MT bundles, known to guide mitochondrial movement (Leterrier et al. 1994). The potential of the method for visualization of MTs over long distances also became evident when imaging MTs at microtubule organizing centers (MTOCs). Figure 4B shows interactions between MTs, a mother centriole, and two matured daughter centrioles. The visualization of MTs in the vicinity of centrosomes was an ideal subject for systematic studies of MW fixation/extraction. Conventionally fixed embedding of PtK2 cells displayed sporadic MTs, which could be considered part of a complex MT aster routinely observed with the light microscope (Figure 5A, arrowheads). The limited visibility of details of the MT aster in conventionally fixed cells was in agreement with previously published results obtained from the same cell line (Baron and Salisbury 1988; Baron et al. 1994). In contrast, MW-fixed cells showed a more complex arrangement of MTs as part of the aster (Figure 5B). Characteristically, the MTs met in crossing points at a well-defined distance from the center of centriole (arrows). Saponin, added to the fixative, led to a loss of pericentrosomal and of more distant, cytoplasmic material. However, it increased the visibility of MTs proximal to the centriole, whereas the centriole itself appeared darker in contrast (Figure 6C). As a result, it became clear that MT asters are not composed exclusively from MTs running radially with respect to the center but also from MTs passing centrioles in short distance tangentially (arrowheads).
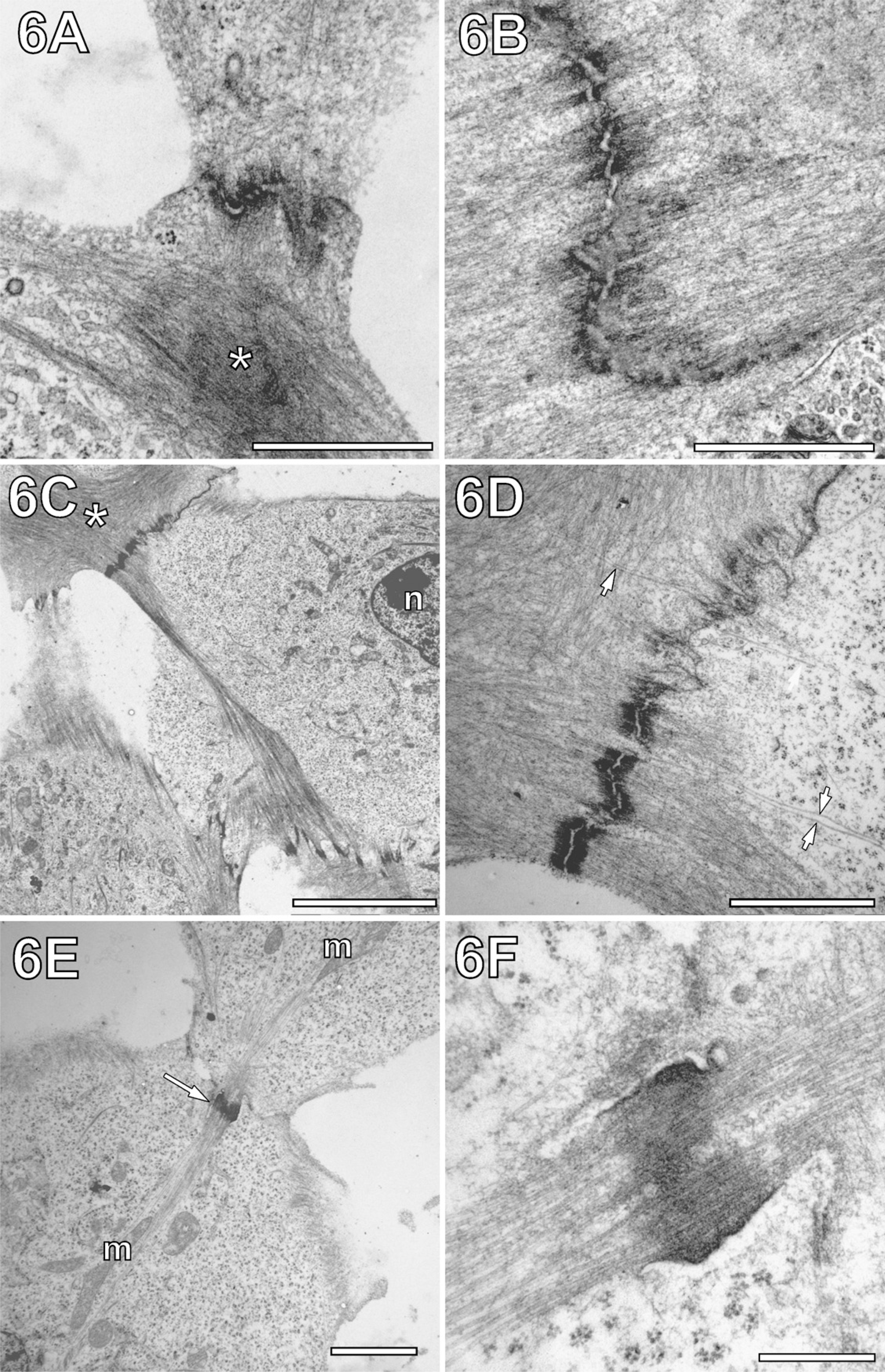
Improved visualization of cell junctions in association with cytofilaments after MW fixation. (
The combination of GA and saponin for MW fixation also led to improved preservation and visualization of cellular junctions and cortical actin. In conventional chemically fixed cells, actin filaments were displayed as a diffuse network, making it difficult to spot individual fibers (Figures 6A and 6B). Sometimes cortical actin seemed to be segregated in electron-dense “clouds” surrounded by network regions of lower electron density (Figure 6A, asterisk). This contrasted with observations in MW-fixed cells, where actin networks of rather homogeneous electron density in conjunction with adherence junctions were found (Figures 6C and 6D). Profound differences in fine structure were also found for the adherence junctions themselves. In conventional chemically fixed cells, adjacent cell membranes formed rather undulated junctions. Their characteristic electron-dense bands at both sides of the cell-cell interface merged quite diffusely with the actin network (Figures 6A and 6B). MW-fixed adherence junctions displayed less undulated cell membranes and electron dense bands, which contrasted sharply with the adjacent actin network (Figures 6C and 6D). As shown in Figure 6D, the combined MW fixation and extraction method enabled not just visualization of the actin network in the form of clearly recognizable individual filaments, but also of MTs approaching the cell junctions, which otherwise would be hidden by cortical actin. Moreover, striking arrangements of MTs of adjacent cells were found in the form of MT-tethering junctions, giving the impression of intercellular MT networking (Figures 6E and 6F).
Discussion
In contrast to bulky tissue samples, cell monolayers of a few micrometers in thickness provide ideal depths for nearly gradient-free fixation. However, to achieve equally high fixation quality for all cells on the cover-slip, in addition, a uniform lateral MW field distribution in the plane of the substratum is needed. Importantly, it turned out that alterations of the field within the MW “hot spot,” temporary fluctuations of the magnetron output, and variations in positioning of the sample (within a radius of ∼2 cm) did not diminish reproducibility of the fixation quality, as long as the number of MW pulses was not less than three pulses.
Our data confirmed previous reports about the suitability of buffered GA for MW-accelerated fixation of cellular fine structures (Wild et al. 1989). Under conventional conditions of chemical fixation, GA cross-linkage is rapidly initiated and continues as a slow reaction that lasts many minutes, if not hours (Rasmussen and Albrechtsen 1974; Griffiths 1993). Here, we applied short, powerful MW pulses for effective cross-linkage already on a subminute time scale. Because excessive heating by MWs could be detrimental for cellular structures, the MW output had to be limited. According to previous publications, sample heating should not exceed 50 ± 5C (Login et al. 1998). A temperature of 50C was given as an upper limit to preserve MTs (Jensen and Harris 1989). Notably, these temperature values refer to the warming up of the fixative, rather than the samples themselves. By adjusting the warming up to the lower range of 35–40C, we took the possibility into account that, locally, sample temperatures may rise above the measured average temperature of the fixative but still stayed below the 50C limit. We believe this empirical measure helped to prevent artifacts typical of prolonged MW exposure (Kok and Boon 1992), while conditions for effective MW-accelerated GA cross-linkage were still maintained at this temperature level.
Using MW ovens other than the EMS-820 may require adjustment of the protocol to MW oven-specific conditions, such as pulse length, warm-up time of the magnetron, and field distribution (for details, see Login and Dvorak 1994). Because operation at a certain output power is realized by timing MW pulses at full power, the real exposure time can differ significantly from the selected processing time. Using the EMS-820, programming of 50% output power and a processing time of 15 sec resulted in three MW pulses, each with a duration of 1.5 sec, at 100% magnetron intensity. Unfortunately, previously published protocols on MW fixation of cell monolayers by Kok and Boon (1992) and McDonald (2002) did not discriminate between the programmed time and the real exposure time. If compared on the basis of the programmed time, we used six to eight times shorter MW exposure. However, by raising the output power 5-fold, to ∼400 W, we achieved a similar thermal effect as reported by Kok and Boon (1992).
To test the strength of the MW-accelerated GA cross-linkage and to seek ways to raise the visibility of cytoskeletal aspects, we challenged our cell monolayers with high concentrations of saponin. Among the options of saponin treatment tested, only a combination of MW-accelerated extraction and fixation in a single step (0.5% GA and 0.5% saponin in 0.1 M cacodylate buffer) showed gradual improvement in visibility of the cytofilaments without signs for severe destruction of membrane-bound organelles, such as rER, Golgi complexes, and mitochondria. From our point of view, the gain in visibility could also be used for systematic studies of ER–mitochondrion interaction as part of Ca2+ homeostasis (Pizzo and Pozzan 2007). However, saponin should be omitted for studies of the MW-fixed fine structure of organelles.
The protocol for MW-accelerated GA fixation presented here could help to bridge the technical gap between rapid cryoimmobilization of cell monolayers and their slow chemical fixation in the conventional way. This would open new opportunities for ultrastructural studies of a number of dynamic processes that video microscopy brings to light. For example, MT-tethering at the cell cortex, occurring at a time scale of minutes, could perhaps be studied using electron microscopic “snap shots” obtained by MW fixation (Ligon et al. 2001; Reipert 2007). Organelle dynamics, such as mitochondrial fission and fusion, could also profit from MW-accelerated immobilization (Bereiter-Hahn and Vöth 1994; Chan 2006). For any future applications, it will be imperative to evaluate the preservation of fine structural details by comparison with cryofixation, the method that is currently regarded as most reliable. Combination of our MW fixation protocol with cryofixation and low-temperature postfixation, dehydration, and embedding in an automatic freeze substitution unit may further improve preservation of the MW-fixed cell monolayers (Murk et al. 2003).
Footnotes
Acknowledgements
S.R. is supported by the Austrian Science Fund (Project P19381).