
Abstract
Recent advances in tissue engineering offer considerable promise for the repair of focal lesions in articular cartilage. Here we describe (1) the macromolecular organization of tissue-engineered neocartilage grafts at light and electron microscopic levels, (2) their in vitro development, and (3) the effect of chondrocyte dedifferentiation, induced by monolayer expansion, on their resultant structure. We show that grafts produced from primary cultures of chondrocytes are hyaline in appearance with identifiable zonal strata as evidenced by cell morphology, matrix organization, and immunohistochemical composition. Like native articular cartilage, their surface zone contains type I collagen, surface zone proteoglycan, biglycan and decorin with type II collagen, aggrecan, chondroitin sulfate, chondroitin-4-sulfate, and keratan sulfate, becoming more prominent with depth. Assessment of cell viability by Live/Dead staining and cell-cycle analysis with BrDU suggest that the in vitro tissue has a high cellular turnover and develops through both appositional and interstitial growth mechanisms. Meanwhile, cell-tracker studies with CMFDA (5-chloromethyl-fluorescein diacetate) demonstrate that cell sorting in vitro is not involved in their zonal organization. Finally, passage expansion of chondrocytes in monolayer culture causes progressive reductions in graft thickness, loss of zonal architecture, and a more fibrocartilaginous tissue histology, consistent with a dedifferentiating chondrocyte phenotype.
Keywords
R
Although this type of therapy has shown significant progress (Brittberg et al. 2003), it has still not fully lived up to expectations (Wood et al. 2006). The reported loss of periosteal/collagen membranes, together with their underlying cells, into the synovial cavity remains an issue; and the development of a fibrocartilaginous repair tissue, which lacks the appropriate organization and material properties to the original hyaline tissue, remains problematic. Additional concerns yet to be resolved include imperfect cartilage resurfacing, unsatisfactory integration of the new tissue with the surrounding matrix, and inadequate assimilation/remodeling of the carrier or scaffold (reviewed in Hunziker 2002; Wood et al. 2006).
Considering some of these problems, an alternative and potentially improved approach would be to engineer a neocartilaginous repair tissue completely ex vivo and then implant the mature construct within the intrachondral lesion site (Kandel et al. 1995). The potential advantages of this approach would be 3-fold: (1) growth conditions would be subject to a high degree of regulation in vitro, otherwise impossible within a diseased or damaged joint; (2) there would be no requirement for a periosteal/collagen flap or carrier because the engineered repair tissue would already possess a mature, fully differentiated cartilage matrix; and (3) this would make the repair and resurfacing of greater and more degenerative lesions a possibility, potentially without the requirement for open knee surgery. Thus, providing the appropriate technologies for implant fixation were in place (Kandel et al. 1995), these potential advantages would translate to an improvement in the scope and success of the repair and, significantly, a reduction in patient rehabilitation time.
Articular cartilage would appear well suited to an ex vivo regeneration strategy of the type described: it is a relatively simple tissue with a low cell-to-matrix volume ratio; it contains a single cell type, the chondrocyte, and it has no innervation, blood, or lymphatic supplies (reviewed by Poole et al. 2001). Yet the superficial simplicity of articular cartilage belies its underlying complexity. The mature tissue is both structurally and functionally heterogeneous and consists of four distinct zones: superficial/tangential, middle/transitional/intermediate, deep/radial, and a zone of calcified cartilage. Each of these zones has striking differences in chondrocyte morphology, gene expression, biochemical composition, and collagen fiber organization (refer to Figure 1). Collectively, these attributes allow the tissue to resist and dissipate biomechanical load while providing smooth, low-friction joint articulation for movement.
Over the last decade there have been a number of interesting publications, chiefly from the Kandel laboratory, describing neocartilaginous tissues engineered, scaffold-free, from high-density primary chondrocytes (Boyle et al. 1995; Kandel et al. 1995, 1997, 1999; Peel et al. 1998; Sun and Kandel 1999; Adkisson et al. 2001; Waldman et al. 2002, 2003a, b, 2004a, b, 2005; Park et al. 2006; Allan et al. 2007). Primarily, cartilage tissues have been fabricated either upon porous, collagencoated synthetic membranes or as biphasic constructs consisting of a cartilaginous tissue grown upon a calcium polyphosphate substrate. Interestingly, the engineered tissues have been shown to possess broad similarities with native articular cartilage and have been used for allogeneic transplantation in animal models with variable success rates (Kandel et al. 1995, 2006; Peel et al. 1998; Park et al. 2006; Allan et al. 2007).
Significant potential exists in the further development of these types of neocartilaginous tissues for the repair of human intrachondral lesions. However, to ensure their successful integration and functioning in the long term, rigorous evaluation of their macromolecular composition and in vitro growth characteristics is clearly essential. Furthermore, to generate sufficient quantities of cells for an autologous human application, harvested chondrocytes would require proliferation in monolayer growth conditions. Crucially, this type of culture induces their dedifferentiation (Benya et al. 1978), characterized by a change in their cellular morphology, a downregulation of cartilage-specific genes (e.g., Sox 5, 6, and 9, aggrecan, and type II collagen), and a reduced ability to produce hyaline cartilage (Stokes et al. 2001; Yang et al. 2006). Whereas chondrocytes can be partially redifferentiated by their transferral to a three-dimensional culture model more conducive toward chondrogenesis, e.g., high density or agarose (Benya and Shaffer 1982; Watt 1988); the subsequent ability of these cells to synthesize genuine hyaline tissue remains ambiguous. Thus, in the present study, we (1) describe the macromolecular composition and organization of scaffold-free neocartilage tissues grown on synthetic filter membranes at light- and electron-microscopic levels, (2) explore their in vitro growth characteristics, and (3) examine the effect of passage expansion of chondrocytes, in monolayer culture, on resultant tissue architecture.
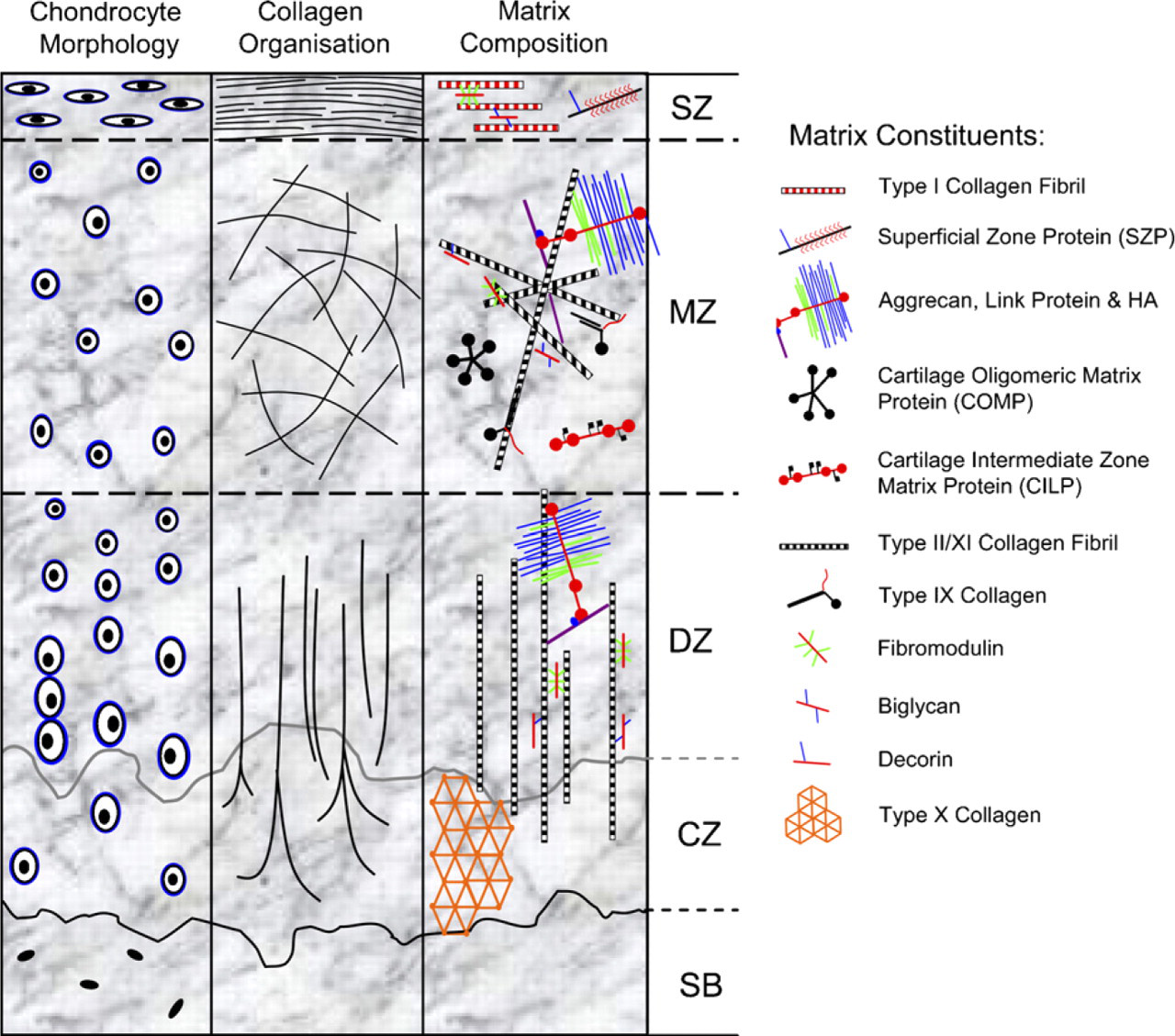
Schematic summarizing the macromolecular organization of mature articular cartilage. The tissue consists of four distinct zones: superficial (SZ); middle (MZ), deep (DZ), and a zone of calcified cartilage matrix (CZ), below which is the subchondral bone (SB). Each zone is distinct in terms of its cell morphology (left panel), collagen fiber organization (middle panel), and the biochemical composition of its extracellular matrix (ECM) (right panel). Matrix constituents of each cartilage zone are presented as molecular schematics.
Materials and Methods
Cell Culture
Full-depth slices of articular cartilage were obtained from the hock joints of 7-day-old bovines under sterile conditions. Cartilage slices were digested in 7 U/ml pronase (Roche; Hertfordfshire, UK) for 3 hr at 37C followed by overnight digestion in 100 U/ml type II collagenase (Worthington Biochemical Co.; Berkshire, UK). To obtain enriched subpopulations of cells from each cartilage zone for tracker studies, the tissue was first subdissected into surface, middle, and deep layers and corresponding layers pooled prior to the above digestion steps. The resultant cell suspensions were filtered through a 40-μm cell strainer (BD Falcon; Erembodegem, Belgium), washed in DMEM containing 4500 mg/liter glucose and L-glutamine (DMEM; Gibco BRL, Paisley, UK), and seeded at high density in 0.5-ml volumes (see below) onto type II collagen-coated (0.5 mg/ml; Sigma-Aldrich, Dorset, UK) 0.6 cm2 Millipore filter inserts (0.4-μm pores; Millipore, Billerica, MA) (Kandel et al. 1995). Initially, to investigate the effect of cell-seeding density on graft histology, chondrocytes were seeded at a range of cell densities: 2, 4, 6, 8, 10, and 12 × 106 cells/insert (i.e., 4, 8, 12, 16, 20, and 24 × 106 cells/ml). To assess the effect of culture time on graft thickness, cells were cultured for 2-, 4-, 6-, and 8-week periods. Subsequently, grafts were made with 6 × 106 cells per insert and typically cultured for 4 weeks. Cultures were fed three times weekly on DMEM containing 20% heat-inactivated FBS, TGF β2 (5 ng/ml; PeproTech, London, UK), and ascorbate (100 μg/ml) and were maintained at 37C in a humidified atmosphere containing 5% CO2 as described by Kandel et al. (1995). To study the effect of dedifferentiation on the histological organization of the graft tissue, chondrocytes were serially passage expanded in monolayer culture in DMEM containing 10% FBS. Briefly, cells were grown to confluency in aerated 75-cm2 flasks before passage with trypsin–EDTA (Gibco BRL). Harvested chondrocytes were washed, counted, and seeded at a density of 6 × 106 cells per filter insert and grown for 4 weeks in culture as described above. Meanwhile, the remaining cells were returned to monolayer culture at low density for further rounds of expansion. This procedure was repeated until passage number five, by which time the growth rate in monolayer was insufficient to sustain sufficient cell numbers for further graft production.
Histology
Cartilage grafts were fixed in 2% paraformaldehyde and processed into paraffin wax using standard histological methods. Six-μm transverse sections were cut through the diameter of the graft and mounted on HistoBond (R. A. Lamb; Eastbourne, UK) slides. Sections were stained for proteoglycan (PG) content in 1% (w/v) alcian blue 8GX (pH 2.5) and Mayers hematoxylin/eosin or for collagen fiber organization by staining in 0.1% (w/v) sirius red F3B in saturated aqueous picric acid. Tissue organization was evaluated using bright-field, differential interference contrast (DIC), and polarizing optics. Graft thickness was measured at 10 arbitrary points across the diameter of the tissue on three different tissue sections, i.e., 30 measurements per graft.
Electron Microscopy
For ultrastructural investigation, grafts were processed for both scanning and transmission electron microscopy. For transmission electron microscopy, tissue was fixed in 2% glutaraldehyde in 0.05 M cacodylate buffer, pH 7.4, containing 0.7% ruthenium hexamine trichloride (RHT; Sigma-Aldrich) for 2 hr, followed by postfixation in 1% OsO4 in 0.1 M cacodylate, pH 7.4, containing 0.7% RHT for 2 hr. Samples were subsequently processed into Spurr's resin (Agar Scientific; Essex, UK) using routine methodology. Ultrathin sections were cut and then stained with uranyl acetate and lead citrate before examination in a Philips EM 208 transmission electron microscope (Philips Electron Optics; Eindhoven, The Netherlands). For scanning electron microscopy, RHT was omitted from the fixatives to prevent PG retention within the tissue. After the primary fixation step, samples were macerated in 10% NaOH for 1 week at 4C to facilitate an appraisal of the collagenous network. After maceration, the tissue was processed for scanning electron microscopy using routine methodology and viewed in a Philips XL-20 scanning electron microscope (Philips Electron Optics).
Immunohistochemical (IHC) Analysis of Graft ECM
Cartilage grafts were fixed in cold 75% ethanol, cryoprotected in PBS containing 5% sucrose, and snap frozen onto cryostat chucks in Cryo-M-Bed tissue mountant (Bright Instruments; Cambridgeshire, UK). Grafts were cryosectioned at 10 μm and frozen sections collected onto Histobond (R. A. Lamb) slides for immunostaining. Sections were labeled by standard indirect immunofluorescence procedures using primary antibodies to a wide range of ECM components (see Table 1). All immunoreagents were diluted in 0.05 M PBS containing 0.1% Tween 20 (Sigma-Aldrich), which was also used for each washing step. Where necessary, either to generate a neo-epitope or to improve antibody penetration, sections were enzymatically deglycosylated by pretreatment with either 0.5 U/ml chondroitinase ABC or chondroitinase B (both from Sigma-Aldrich) and 0.5 U/ml keratanase (AMS Biotechnology; Oxon, UK) in 100 mM Tris–acetate buffer (pH 7.4) for 1 hr at 37C (see Table 1). After washing, sections were treated with blocking serum (Dakopatts Ltd.; High Wycombe, UK) at 1:20 dilution for 30 min at room temperature before overnight incubation at 4C with primary antibody (see Table 1). After a second wash, sections were fluorescently labeled with FITC-conjugated goat anti-mouse or swine anti-rabbit Fab fragments (Dakopatts Ltd.), recognizing either mouse or rabbit species, for 30 min at room temperature. Sections were counterstained with propidium iodide (0.5 μg/ml; Molecular Probes, Invitrogen, Paisley, UK) for 5 min to impart nuclear context and then mounted in Vectashield faderetarding mountant (Vector Laboratories Ltd.; Peterborough, UK). Control sections were incubated with PBS, 10 μg/ml mouse immunoglobulins (Sigma-Aldrich), or non-immune rabbit serum (Sigma-Aldrich) instead of primary antibody, with or without the enzymatic digestion step. Sections were viewed and photographed using a Leitz fluorescent microscope (Leica; Wetzlar, Germany) equipped with digital image acquisition.
Antibodies used for immunohistochemistry
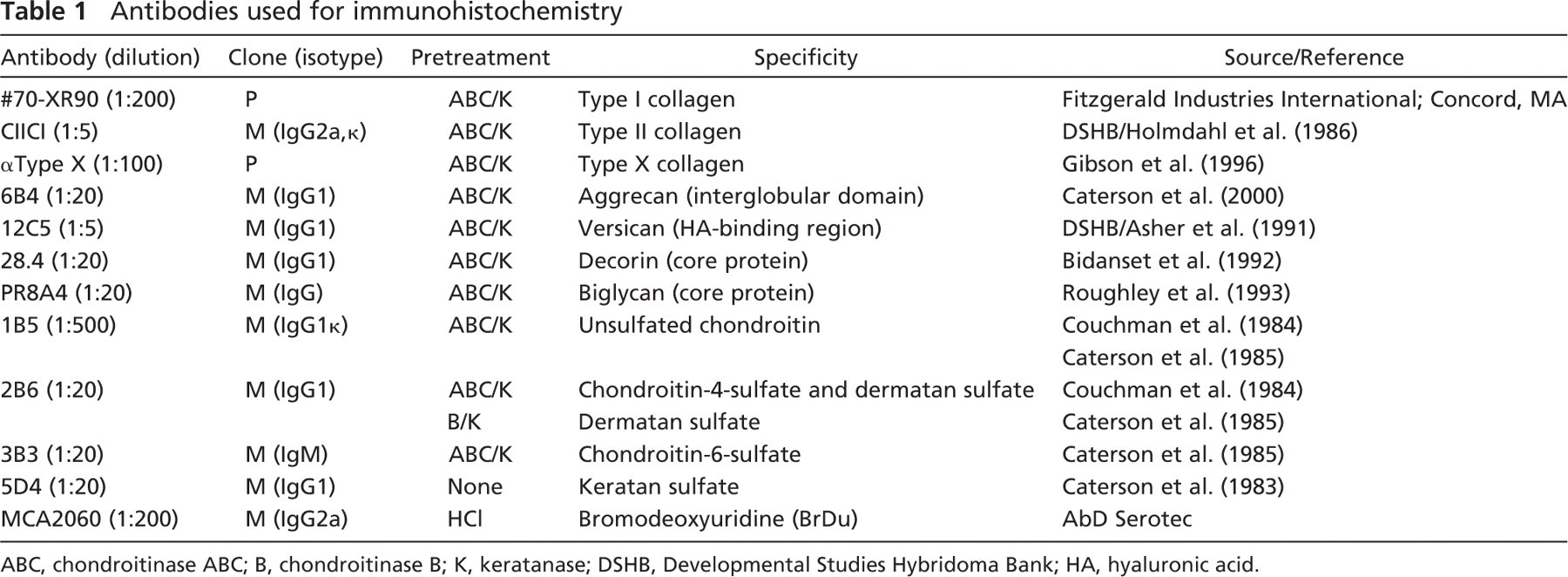
ABC, chondroitinase ABC; B, chondroitinase B; K, keratanase; DSHB, Developmental Studies Hybridoma Bank; HA, hyaluronic acid.
Cell Viability Assay
To determine cell viability and identify the presence and distribution of dead cells within the neocartilage tissue, grafts were incubated in the presence of 2 μm calcein AM and 4 μM ethidium homodimer solution (Live/Dead Viability/Cytotoxicity Kit; Molecular Probes, Invitrogen) for 30 min. After incubation with the above reagents, grafts were washed in PBS, cut transversely into thin (∼0.25 mm) slices using a sharp razor blade to minimize cell death, and mounted under coverslips in PBS. Sections were photographed immediately under epifluorescence optics as described previously.
Cell Proliferation
To identify proliferating cells and study the role of cell proliferation on tissue reorganization, neocartilage grafts were incubated with bromodeoxyuridine (BrDU, 1:1000; Amersham, Buckinghamshire, UK) in complete tissue culture medium for 90 min at 37C. Grafts were then fixed and processed for paraffin wax histology as described previously. A rat monoclonal antibody toward BrDU (AbD Serotec; Oxford, UK) (see Table 1) was used to localize proliferating cells using immunoperoxidase labeling in conjunction with DAB staining. This procedure was, in essence, similar to that described previously with the following modifications. After sections had been dewaxed and rehydrated they were incubated in 1 MHCl at 60C for 10 min to denature the DNA and improve antibody access. Endogenous peroxidase labeling was then irreversibly inhibited using 1.5% hydrogen peroxide (Sigma-Aldrich) in 100% methanol for 1 hr, followed by washing in PBS prior to the antibody blocking stage using rabbit serum (1:20). Sections were incubated with the anti-BrDU antibody (1:100) for 1 hr, washed in PBS, and then treated with a rabbit anti-rat biotinylated secondary antibody (1:200; Vector Laboratories Ltd.) for 30 min at room temperature. To check for nonspecific labeling with primary and secondary antibodies, respectively, tissue sections were incubated with either naive rat immunoglobulin or antibody diluent (i.e., PBS) instead of the anti-BrDU antibody. After washing in PBS, sections were incubated with avidin/biotin/peroxidase complex (ABC; 20 μl avidin DH, 20 μl biotinylated enzyme, and 1 μl PBS) for 30 min. The ABC complex was localized using a DAB kit (Vector Laboratories Ltd.) including nickel chloride in the reaction substrate to produce a black/brown reaction product. As a further check for nonspecific staining, the ABC complex or DAB staining steps were omitted. Sections were counterstained with 1% (w/v) alcian blue 8GX (pH 2.5) to impart tissue context and then photographed using bright-field optics.
Cell-Tracker Studies
To follow the in vitro kinetics of graft development, enriched chondrocyte subpopulations from each cartilage zone were labeled with a green fluorescent cell-tracker probe (Vybrant CFDA cell tracer kit; Molecular Probes, Invitrogen). To determine the extent of interzonal cell movement occurring during culture, a labeled mid-zone subpopulation was layered between unlabeled deep- and surface-zone fractions, allowing 3 hr between the addition of successive cell subpopulations to minimize the extent of initial cell mixing. Additionally, to identify the potential for zonal cell sorting during the culture period, labeled surface-, middle- or deep-zone cells were first mixed with their unlabeled counterparts before seeding into filter inserts at a density of 6 × 106 cells per insert (i.e., 2 × 106 cells from each layer). After 4 weeks of culture the grafts were harvested, fixed, and processed into paraffin wax, as described previously. Wax sections were cut and counterstained with propidium iodide (0.5 μg/ml; Molecular Probes, Invitrogen) to impart nuclear context to the tissue or were immunofluorescently labeled with a monoclonal antibody toward chondroitin sulfate to allow visualization of the ECM.
Results
Primary Culture
Macroscopically, all neocartilage graft tissues produced in this study had a glassy, hyaline appearance (Figure 2A). Based upon histological, ultrastructural, and IHC criteria, the grafts were stratified into well-defined cartilaginous zones that approximated with the superficial, middle, and deep layers of mature articular cartilage, although no calcified zone was identifiable (see below). In primary culture, this histological zonal organization was apparent at all cell-seeding densities and all culture periods studied (Figure 2, Figure 3, and Figure 4).
Cell Density and Culture Duration
Cell-seeding density and culture period were important factors in determining the thickness of the resultant neocartilage tissues. Increasing cell number from 2 to 4 × 106 yielded a highly significant increase (p<0.001) in the thickness of neocartilage grafts produced at all culture periods examined. Increasing cell number >4 × 106 cells per insert provided subtle increases of variable significance in resultant graft thickness (Figures 2B and 2C). Consistent with an increase in cell-seeding density was a concomitant increase in graft tissue cellularity; thus, seeding >6 × 106 cells per insert yielded a tissue with a high cell-to-matrix ratio characterized by an excessively hypercellular appearance. The effect of increasing the duration of culture from 2 to 8 weeks was a steady increase in the thickness of the cartilage grafts produced at all cell-seeding densities (Figures 2B and 2C). This appeared to result from a combination of cell proliferation, hypertrophy, and progressive accumulation of ECM material (see below). Interestingly, at each time point examined, the zonal proportions of the graft appeared relatively constant, suggesting that in vitro growth occurred uniformly throughout the tissue during culture. Furthermore, with advancing time the cells in the basal part of the tissue (i.e., those closest to the filter membrane) appeared to acquire a more columnar organization (Figure 2C; see below).
Graft Organization
Light and electron microscopic examination of neocartilaginous grafts showed the surface zone to be weakly alcianophilic (Figure 3A), consisting of discoidal cells surrounded by a sparse, fibrillar meshwork of collagen (Figures 3A, 3B, and 3E). In this zone, both cells and matrix had a prominent horizontal organization that was clearly apparent under Nomarski DIC and polarizing optics (Figures 3H and 3I). The underlying mid-zone was strongly alcianophilic, indicating a higher PG content than the surface zone, and contained spheroidal chondrocytes that were separated by significant amounts of ECM (Figures 3A, 3C, and 3F). Ultrastructural observation of this zone showed that its ECM comprised pericellular, territorial, and interterritorial matrix compartments, each with distinct collagen fiber densities and organizations, similar to that of articular cartilage (Figures 3C and 3F). The subjacent deep zone of the tissue was more cellular in appearance than the overlying cartilage. Chondrocytes in this zone had an ovoid, hypertrophic morphology and were densely packed together, with some cells occurring in columnar arrays separated by thin septa of alcianophilic ECM (Figure 2C, Figures 3A, 3D, and 3H). The interterritorial matrix of these septa, evident at the ultrastructural level, consisted of prominent longitudinal fibrils that were arranged vertically with respect to the graft surface (Figures 3D and 3G). Observation with both Nomarski DIC and polarizing optics indicated that tissue organization in the deep zone (Figures 3H and 3J) was perpendicular to that occurring at the graft surface (Figures 3H and 3I).
Composition of Graft ECM
Negative controls showed no nonspecific labeling with either primary or secondary antibodies (data not shown). Immunofluorescent labeling patterns obtained with antibodies toward the chief ECM components suggested that after 4 weeks of primary culture the graft tissue was immunohistochemically similar to immature native articular cartilage (refer to Figure 1).
Collagens. Type I collagen was strongly detectable at the surface of the graft but was absent from the underlying connective tissue (Figure 4A) as occurs within the superficial zone of articular cartilage, whereas type II collagen was identifiable throughout the graft matrix (Figure 4B), confirmatory of the tissue's hyaline nature. Type X collagen was not detectable in grafts after 4 weeks of culture, reflecting the immaturity of the ECM; however, it became weakly associated with the pericellular matrix of hypertrophic chondrocytes in the basal zone after extended culture periods (i.e., >12 weeks; data not shown); its location corresponding to that occurring in articular cartilage.
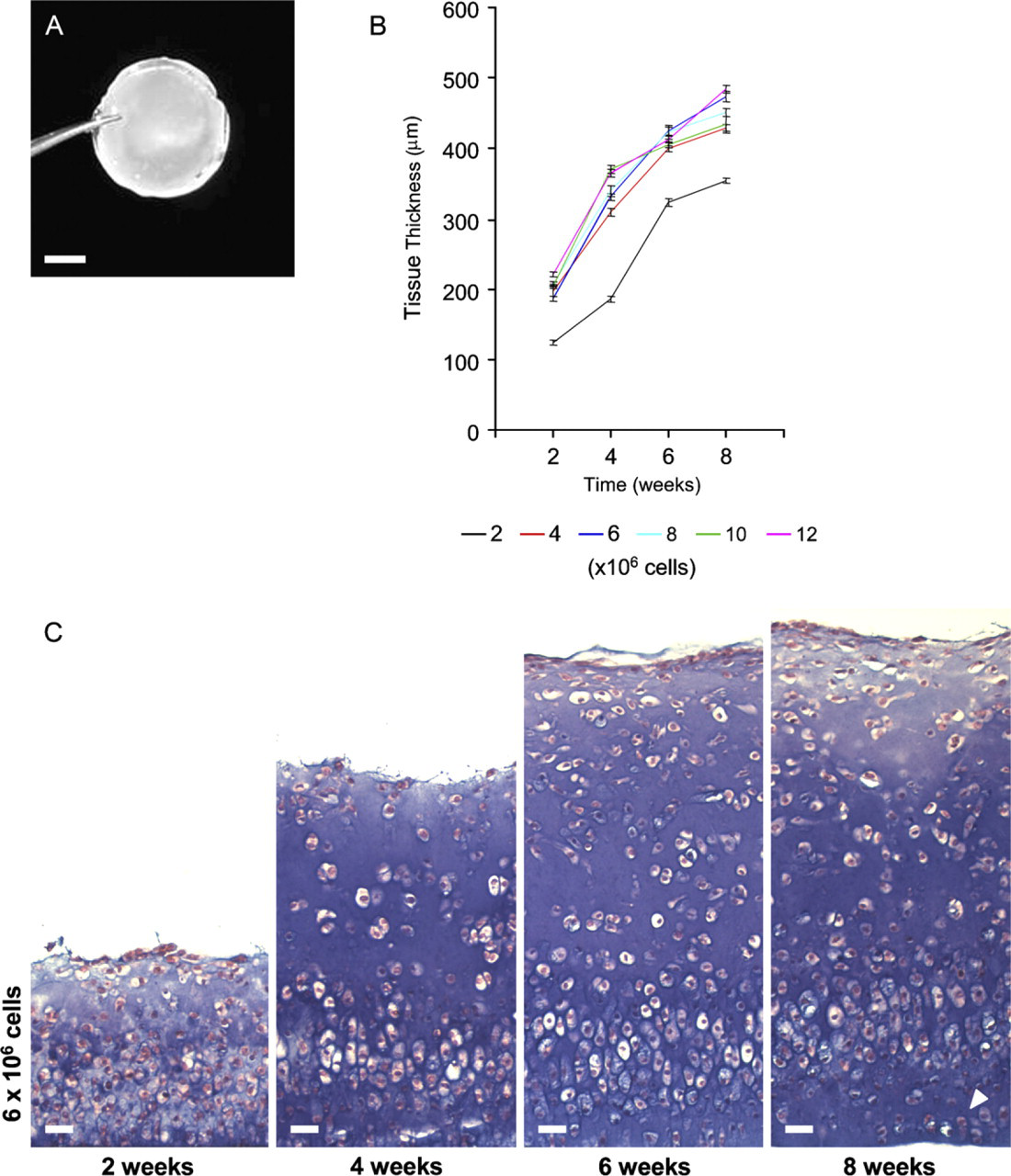
Effect of culture time and cell-seeding density on neocartilage tissue histology (primary culture). (
PGs. Aggrecan, the large aggregating PG of cartilage, was detectable throughout the ECM of the graft tissue (Figure 4C); however, versican was not evident (Figure 4D). The small leucine-rich PGs biglycan and decorin were also detectable throughout the graft tissue (Figures 4E and 4F, respectively), but decorin, in particular, was more prominent in the surface zone of the tissue and diminished with depth, as occurs in articular cartilage. Superficial zone proteoglycan (SZP/lubricin/PRG4/MSF precursor protein) was weakly immunolocated to cells of the graft surface after 4 weeks of culture (Figure 4G); however, with advancing culture time (>12 weeks) it became more prominent at the surface of the graft (Figure 4H), having a similar organization to that of articular cartilage.
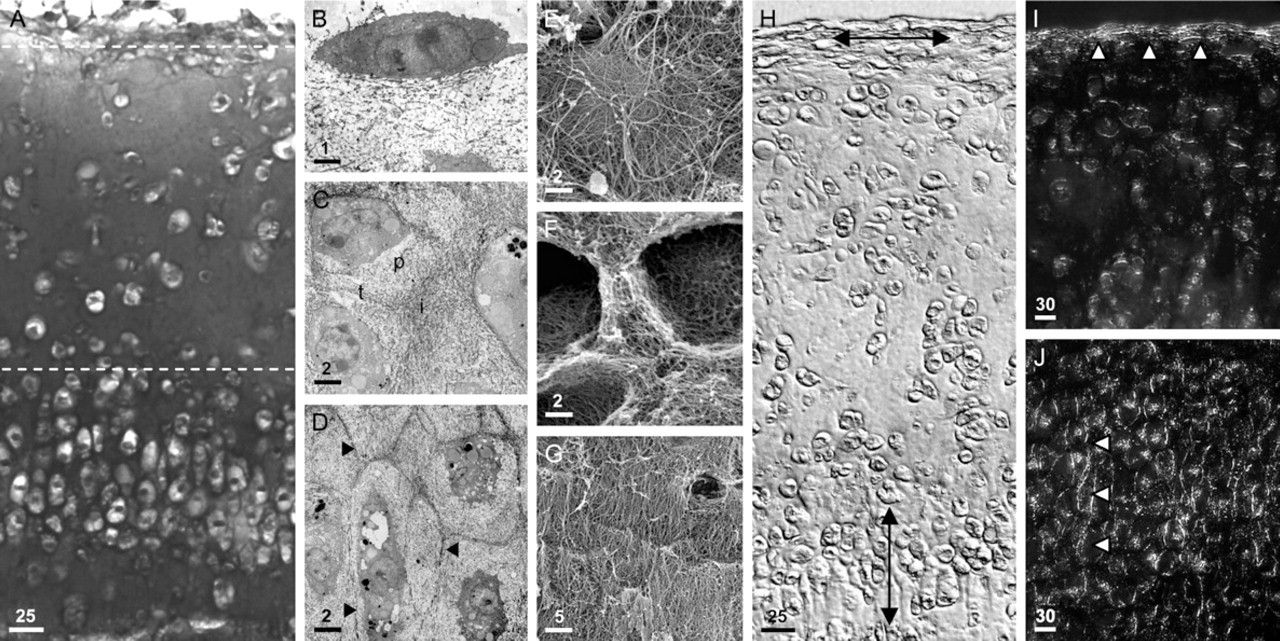
Histological and ultrastructural organization of neocartilage tissue. (
Glycosaminoglycans (GAGs). Unsulfated chondroitin (C-0-S) and C-4-S “stubs” (i.e., after chondroitinase digestion) were prominent throughout the graft ECM and had a similar distribution to aggrecan, suggestive of their association with this PG (Figures 4I and 4J, respectively). Chondroitin-6-sulfated “stubs” were not detectable in the graft ECM (Figure 4K), indicative of the immaturity of the engineered tissue. Keratan sulfate was weakly present in the pericellular matrix compartment, becoming more prominent with depth (Figure 4L) as occurs in articular cartilage, where it is associated with aggrecan. In contrast, dermatan sulfate was present predominantly in the surface zone of the graft tissue (Figure 4M), suggesting that this molecule was associated with decorin that was similarly located and to which it has known association in articular cartilage.
In Vitro Growth Characteristics
Assessment of cell viability with a fluorescent Live/Dead Viability/Cytotoxicity Kit (Molecular Probes, Invitrogen) showed the majority of cells in thin (∼0.25 mm) living sections of graft tissue were viable, i.e., had stained green after 4 weeks of culture (Figure 5A). During the same time period, dead cells (both necrotic and apoptotic) evident by their red nuclei had accumulated throughout the graft tissue; however, there was no distinct zone of cell death per se. Cell-cycle analysis with BrDU revealed strong incorporation of this thymidine analog in proliferating cells both at the graft surface and in the underlying cartilage tissue (Figure 5B), the high levels of BrDU incorporation probably reflecting the known effect transforming growth factor has on chondrocyte proliferation. Comparison with control sections incubated with naive immunoglobulin or antibody diluent instead of BrDU antibody confirmed that there was no nonspecific labeling (data not shown). Cell-tracker studies with CMFDA (5-chloromethyl-fluorescein diacetate) showed that after 4 weeks of culture the labeled mid-zone subpopulation largely retained its intermediate position in tri-layered composites, indicating there was little interzonal movement of chondrocytes (Figure 5C). When grafts were made by mixing a single fluorescently labeled subpopulation with its unlabeled counterparts from the adjacent zones, in all cases the labeled chondrocytes were randomly distributed throughout the tissue depth (Figures 5D–5F). Although there was no compelling evidence of zonal cell sorting mechanism per se, occasionally there appeared to be slightly more labeled deep zone cells in the basal part of the tissue (Figure 5F). This was possibly due to the hypertrophic cells of the deep zone being larger (mean cell volume ± SEM of 1689 ± 145 μm3) and therefore settling more readily during the initial cell-seeding step than the comparatively smaller cells of the middle and surface zones (mean cell volumes ± SEM of 567 ± 14 μm3 and 467 ± 8 μm3, respectively).
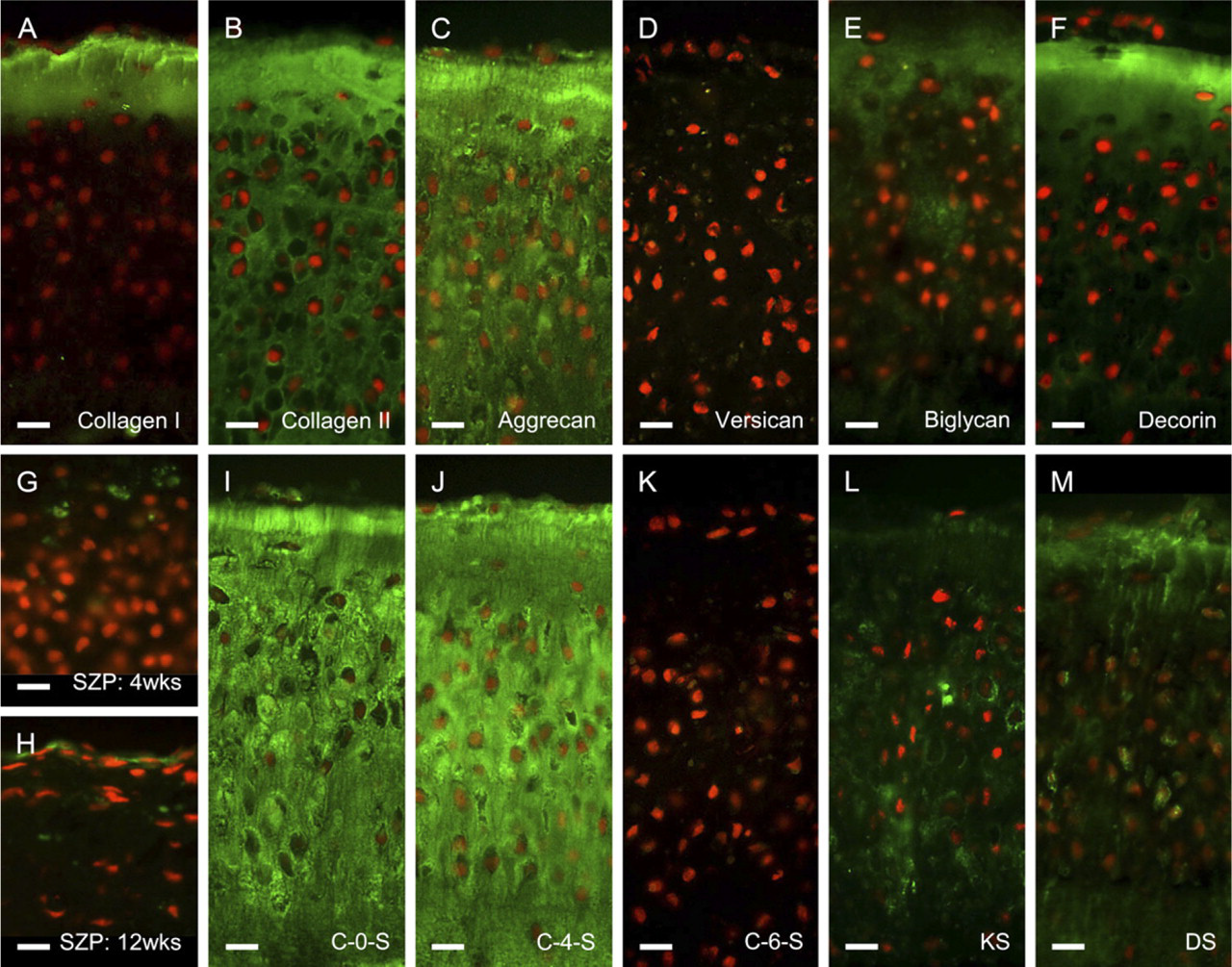
Immunohistochemical composition of neocartilaginous graft ECM. Sections have been labeled (green) with a panel of antibodies toward the chief structural components of the extracellular matrix, i.e., collagens (
Effect of Monolayer Expansion
Successive passage expansion of chondrocytes in mono-layer conditions led to striking changes in tissue depth, histological organization, and IHC composition of the resultant tissue (Figure 6–Figure 8). There were significant deceases in graft thickness from >500 μm at P0 to <300 μm at P5 and a steady reduction in the extent of alcian blue staining after successive passage, indicative of a reduced sulfated GAG content (Figures 6A and 6B). Furthermore, histological organization of the tissue became less apparent with a progressive loss of zonal architecture (Figure 6B). At P3, chondrocytes had a profoundly reduced ability to lay down ECM, resulting in a progressive loss of cells to the medium and premature termination of the cultures at this stage. Interestingly, this capacity was restored following successive passage of cells, although the resultant grafts lacked any semblance of zonal organization, contained discrete clusters of cells in the basal portion of the tissue, and appeared histologically similar to fibrocartilage (Figure 6B). IHC labeling of the grafts also showed a qualitative increase in the extent of labeling for type I collagen and versican and an overall decrease in the extent of aggrecan immunolabeling with increasing passage number (Figure 7 and Figure 8), also indicative of a switch to a more fibrous cartilage phenotype. The labeling pattern for versican was particularly interesting because it coincided spatiotemporally with the cell clusters at the base of the tissue after P3, clearly suggestive of a fibrochondrocytic cell phenotype in this tissue zone.
Discussion
Tissue Organization
This study adds to previous data (e.g., Kandel et al. 1995) demonstrating that chondrocytes grown at high density upon porous filter membranes can produce zonally stratified neocartilage grafts potentially useful for the biological repair of intrachondral lesions of articular cartilage. All tissues engineered from a primary chondrocyte source in this study showed histological, ultrastructural, and IHC similarities to native articular cartilage. The neocartilage tissues comprised three histologically distinct zones that resembled the superficial, middle, and deep zones of native articular cartilage, and within these zones there were striking similarities in cell morphology, collagen fiber organization, and matrix composition to that of native articular cartilage. Interestingly, whereas the histological organization of the tissue closely resembled that of mature articular cartilage, the composition of its ECM suggested closer similarities with immature, fetal-like hyaline cartilage: there was no detectable chondroitin-6-sulfate; type X collagen was initially absent, and there was no calcified cartilage matrix. This was not altogether surprising, however, given that the matrix was synthesized de novo over a short, 4-week period. Furthermore, although we did not specifically quantify the mechanical strength of the tissue, the pliability of the neocartilage evident during handling indicated that it lacked the mechanical stiffness of mature articular cartilage.
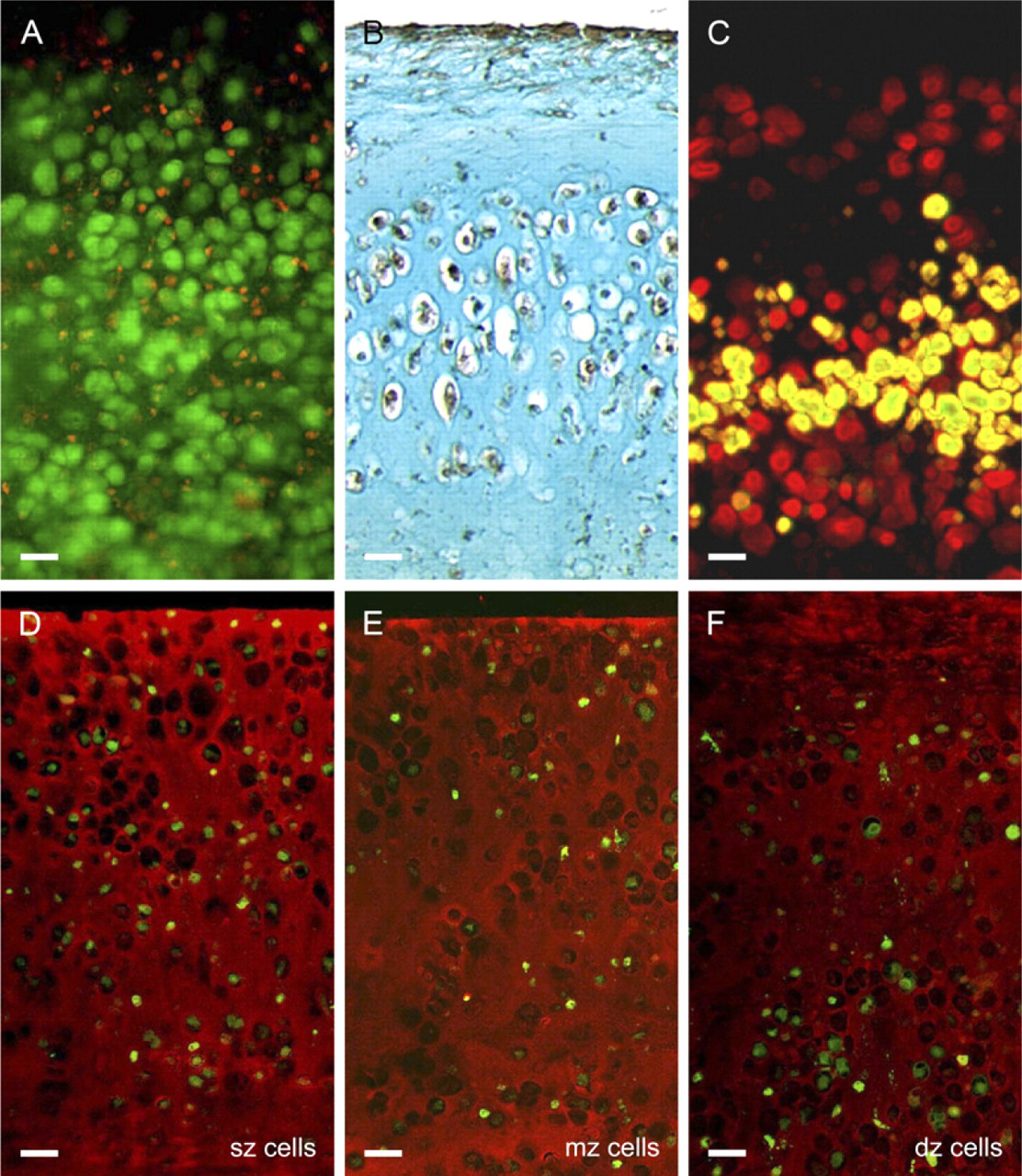
In vitro growth characteristics of neocartilaginous grafts. (
Previous studies, chiefly by Waldman and colleagues, indicate that media supplementation and/or mechanical stimulation have the potential to add functional value to neocartilage grafts. Media supplementation has been successfully used to improve the quality of tissue-engineered cartilage, resulting in a more mature tissue phenotype. β-glycerophosphate, for example, induces deep zone chondrocytes to calcify their cartilage matrix (Kandel et al. 1997; Waldman et al. 2002), and NaHCO3 supplementation has been shown to improve the histological quality of neocartilage, resulting in more flattened cells resembling superficial zone chondrocytes at the graft surface (Waldman et al. 2004a). Biomechanical loading regimes, both compressive and shear (long and short term) have also been used to increase matrix deposition, improve matrix composition, and increase the thickness and biomechanical properties of the engineered cartilage (Waldman et al. 2003b, 2004b, 2005). Indeed, the importance of utilizing appropriate biomechanical kinematics in articular cartilage engineering cannot be overstated. For example, recent data show that chondrocytes require stimulation by applied surface motion, but not axial compression, to upregulate production of SZP and to generate a normal articular surface (Grad et al. 2005). Provision for these factors may thus be important in optimizing the functional value of neocartilage tissues generated for repair purposes. It is worthwhile considering, however, that although an engineered construct with similar mechanical properties to mature articular cartilage may be desirable from a biomechanical standpoint, an immature tissue phenotype may facilitate better integration with the host tissue and could be mechanically conditioned in situ, i.e., postimplantation, via a prudent exercise regimen.
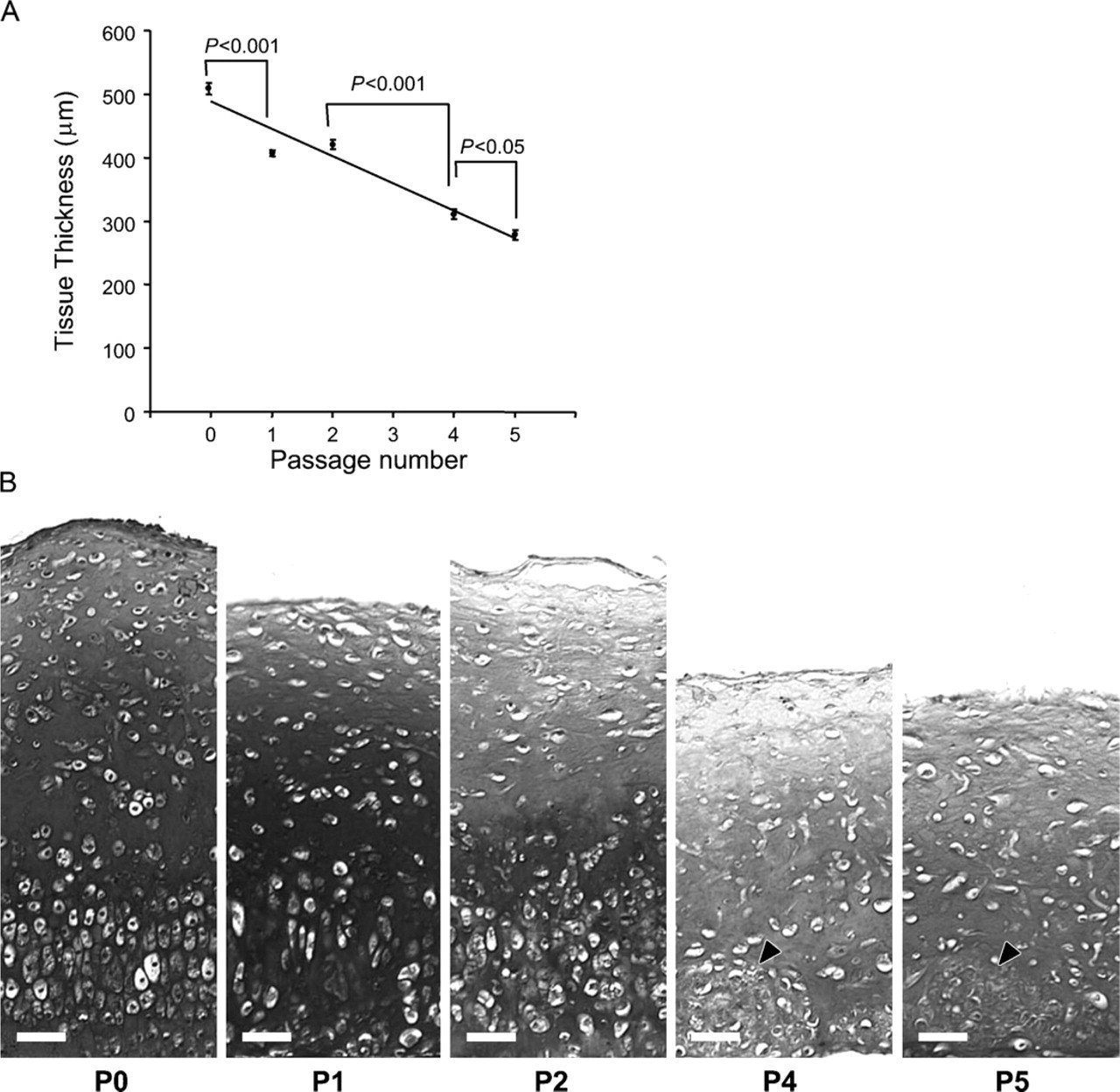
Effect of monolayer expansion upon the histological organization of engineered constructs. (
Cellular Growth Mechanisms
Together our data suggest that the engineered tissues have a high cellular turnover and develop through a combination of appositional and interstitial growth mechanisms. Appositional growth would appear to occur principally at the graft surface, as evidenced by prominent BrDU incorporation at this site, and interstitial growth in the underlying cartilage tissue via a combination of cell proliferation and matrix accumulation. Our cell-tracker studies strongly suggest that zone-specific cell sorting does not play a role in the establishment of the distinct strata in vitro. Instead, our data suggest that chondrocytes divide or differentiate according to their potential and position in the developing tissue and that these behaviors are dictated largely by the culture environment. At the surface of the graft, chondrocytes would begin to flatten out, proliferate, and undergo dedifferentiation, as occurs in monolayer culture (Benya et al. 1978; Stokes et al. 2001). These changes might not only explain the change in cell morphology from spheroid to discoid, but also account for the high levels of BrDU incorporation in cells at the surface of the tissue. It would also drive the expression of molecules typically associated with the superficial zone, such as collagen type I (Benya et al. 1978). Additionally, as the cells spread and assume a more fibroblastic morphology, collagen fibrils would be secreted more across the surface of the graft tissue, as occurs in monolayer culture. Deeper within the tissue the spheroid/ovoid chondrocyte morphology would be preserved as the cells are constrained at high density in three dimensions by neighboring cells. This environment would be conducive toward chondrogenesis (Watt 1988) as reflected in the presence of increased amounts of sulfated PG, type II collagen, and aggrecan deeper within the tissue, potentiated further by the presence of transforming growth factor in the culture medium. During the initial period of growth, effete chondrocytes are probably crushed by the growing tissue and undergo necrosis/apoptosis as evidenced by the red nuclear staining seen with ethidium homodimer-1. Progressive deposition and accumulation of ECM between stacks of densely packed cells at the base of the culture might also generate the interstitial septa seen in this zone, as occurs in the developing growth plate (Eggli et al. 1985). Also, because the tissue is constrained laterally by the walls of the culture vessel, growth would proceed in a predominantly vertical direction, made manifest as an increase in tissue thickness as seen in this study.
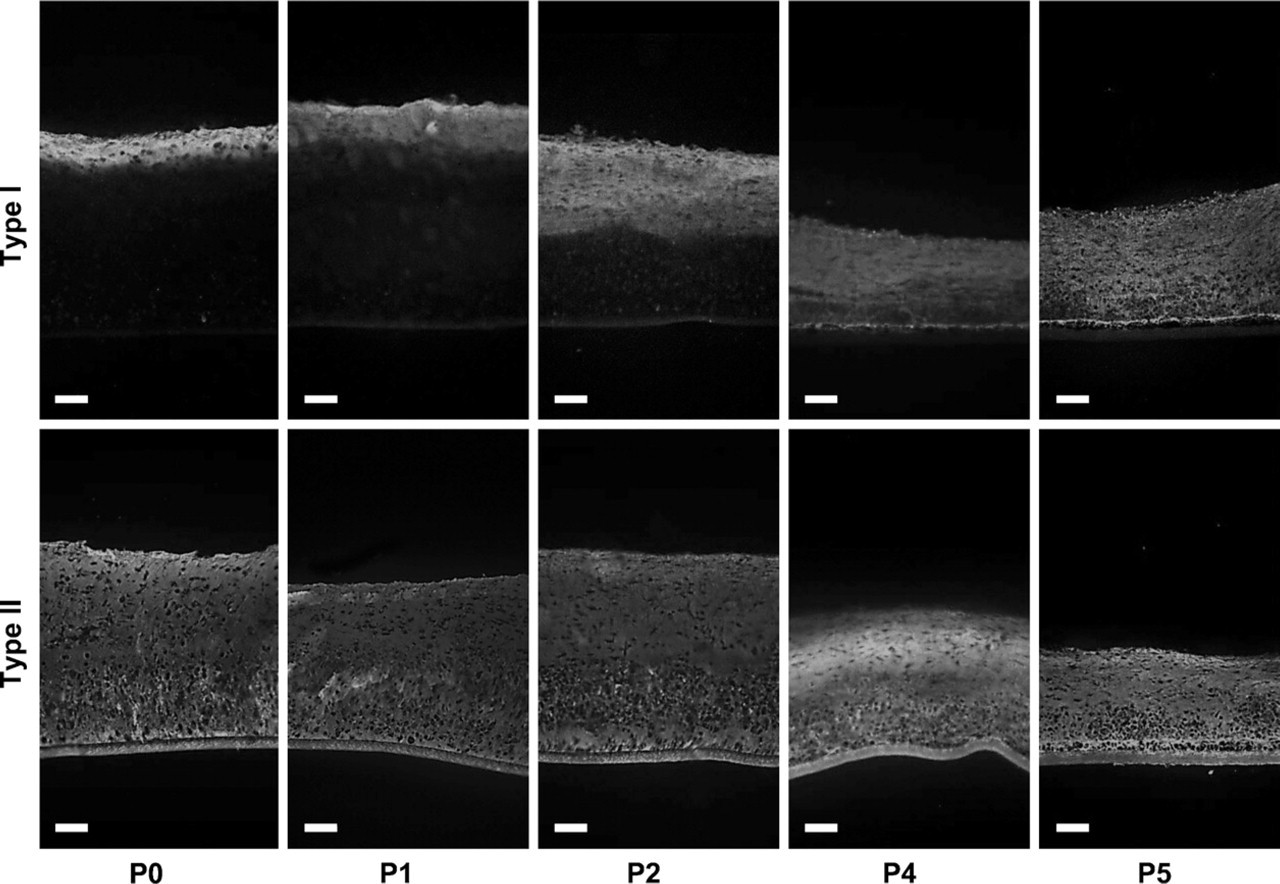
Effect of monolayer expansion upon the distribution of collagen types I and II in neocartilaginous tissue. Note progressive increase in immunolabel for type I collagen with successive passage (P). Qualitative changes in the distribution of type II collagen were not apparent. Bar = 80 μm.
It is conceivable that the distinct strata might also arise partly through metabolic/morphogenic gradients occurring within the developing tissue. These might arise spontaneously as a result of a differential between the metabolic conditions cells experience at the surface of the developing tissue and its base. For instance, at the base of the culture competition for metabolites would probably be greatest because, in this region, cells are most densely packed and depend upon diffusion of metabolites through the underlying filter membrane and the overlying cells and matrix. It is known that oxygen tension, for example, plays an important regulatory role during cartilage growth by promoting chondrocyte differentiation and cartilage matrix synthesis while suppressing terminal chondrocyte differentiation (Hirao et al. 2006). Oxygen tension may play a similar role in the current context: its physiological effects manifesting in the zonal organization of cells and matrix within the in vitro-generated tissue.
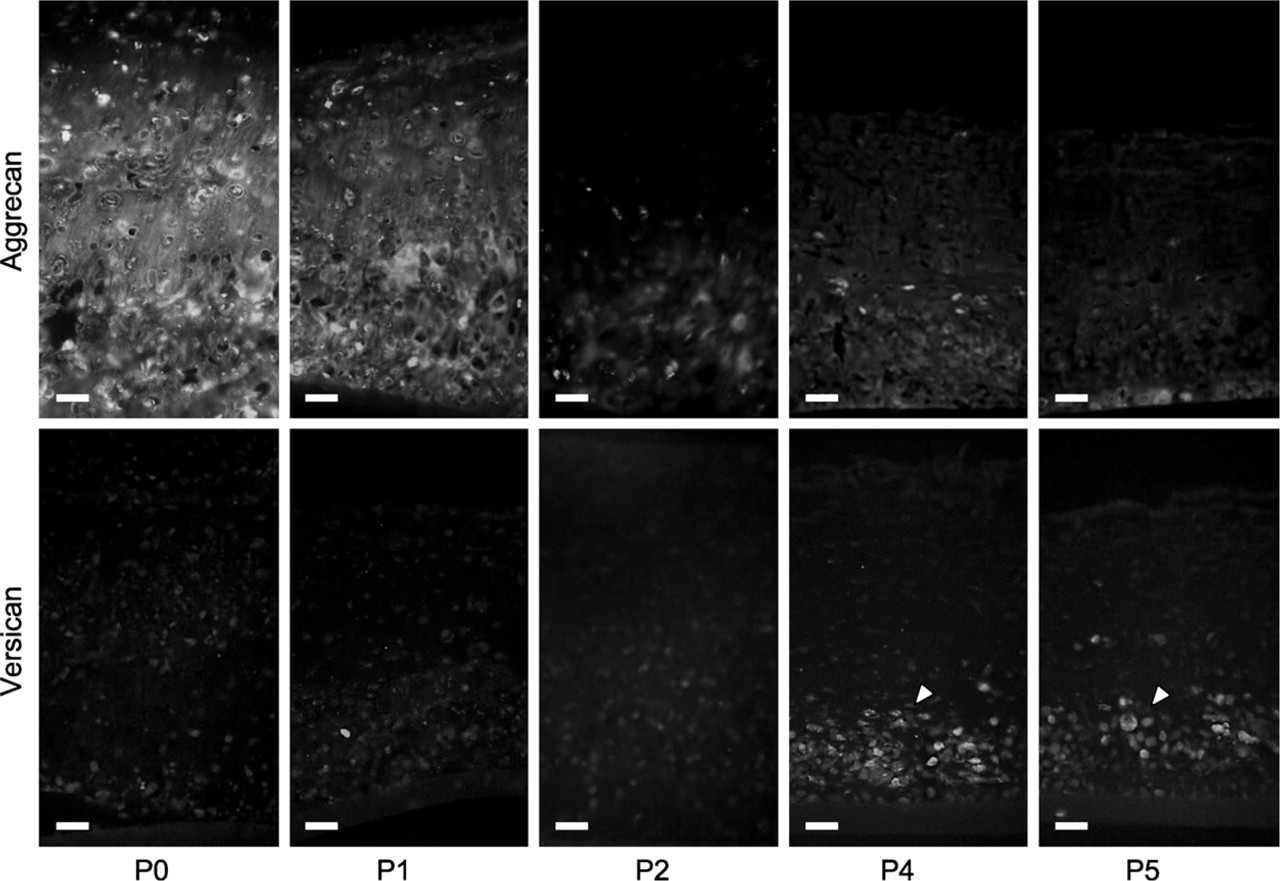
Effect of monolayer expansion upon the distribution of aggrecan and versican in neocartilaginous tissue. Higher magnifications showing progressive decrease in immunolabel for aggrecan with successive passage and corresponding increase in versican immunolabel, particularly associated with cell clusters in the basal part of the tissue after P3 (arrowheads). Bar = 40 μm.
Effect of Passage Expansion
Although chondrocytes are readily obtainable from animal models to generate allogeneic grafts for experimental purposes, to obtain sufficient cell numbers for a clinical application would necessitate multiple expansions of autologous chondrocytes in monolayer culture. It is well established that this type of culture causes their cellular dedifferentiation to a more fibroblastic phenotype: downregulation of cartilage-specific genes such as Sox-5, 6, and 9, aggrecan, and type II collagen and upregulation of type I collagen (Benya et al. 1978; Stokes et al. 2001; Yang et al. 2006). Although there is some evidence that passage-expanded chondrocytes can partially redifferentiate when transferred to culture models more conducive to chondrogenesis (e.g., agarose or high-density cultures) (Benya and Shaffer 1982; Watt 1988), in this study we have shown that their ability to produce true hyaline tissue resembling articular cartilage becomes dramatically reduced.
Other studies have shown similar phenotypic changes to occur in human articular chondrocytes between P0 and P4 in monolayer culture, with profound changes identifiable at the mRNA level identifiable as early as P1 (Darling and Athanasiou 2005). Chondrocytes isolated from human nasal septum cartilage have been shown to exhibit a similar impairment in their ability to proliferate at P3, whereupon they begin to acquire a fibroblastic dedifferentiation morphology (Chua et al. 2005). Binette et al. (1998) have also noted a similar downregulation of mRNA for aggrecan and type II collagen and an upregulation of versican and collagen type I by human articular chondrocytes after monolayer expansion, consistent with the IHC data presented in this study.
Thus, at present, although significant potential exists for the use of autologous chondrocyte grafts for cartilage repair purposes, the deleterious effects of chondrocyte dedifferentiation resulting from multiple passages still remain a major obstacle. Chondrocytes differentiated from stem cells would clearly obviate the necessity to perform multiple expansions of primary cells; thus, stem cell technology currently offers considerable promise. A number of recent studies, for example, have identified chondroprogenitor/stem cell populations within the superficial zone of articular cartilage (e.g., Dowthwaite et al. 2004). These cells have been shown to be highly proliferative, exhibit phenotypic plasticity in their differentiation pathway, and can undergo multiple rounds of passage without loss of chondrogenic phenotype (Dowthwaite et al. 2004; Martin et al. 2005), showing significant potential for future cartilage-engineering applications.
In summary, results of this study add to previous data demonstrating that high-density primary cultures of full-depth chondrocytes grown on Millipore filter membranes produce a zonally stratified hyaline cartilage tissue with histological, ultrastructural, and IHC features of immature articular cartilage. These features arise spontaneously in primary culture and are progressively lost after chondrocytes have been passage expanded in monolayer growth conditions. The resultant changes in graft thickness, organization, and composition thus reflect the cellular phenotypic changes that occur through chondrocyte dedifferentiation. Thus, significant potential exists in the continued development of autologous chondrocyte grafts for the repair of intrachondral lesions of human articular cartilage. There is, however, a pressing need to accommodate recent advancements in cartilage stem cell technology (Dowthwaite et al. 2004), new defined growth medium (Jakob et al. 2001; Chua et al. 2005), cell culture methodologies (Murphy and Polak 2004; Barbero et al. 2006), and gene therapy (Tew et al. 2005) that will allow bulking of chondrocyte numbers without the deleterious effects of cellular dedifferentiation.
Footnotes
Acknowledgements
Monoclonal antibodies CIICI (Holmdahl et al. 1986) and 12C5 (![]() ) were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA. Antibodies toward type X collagen and decorin were generously provided by Professor G. Gibson and D. Bidanset, respectively. We would also like to acknowledge Dr. Mari Nowell for help with data analysis.
) were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA. Antibodies toward type X collagen and decorin were generously provided by Professor G. Gibson and D. Bidanset, respectively. We would also like to acknowledge Dr. Mari Nowell for help with data analysis.