
Abstract
Reproducible visualization of neurons and glia in human brain is essential for quantitative studies of the cellular changes in neurological disease. However, immunohistochemistry in human brain specimens is often compromised because of prolonged fixation. To select cell lineage–specific antibodies for quantitative studies of neurons and the major types of glia, we used 29 different antibodies, different epitope retrieval methods, and different detection systems to stain tissue arrays of formalin-fixed human brain. The screening pointed at CD45/leukocyte common antigen (LCA), CD68(KP1), 2′, 3′ cyclic nucleotide phosphatase (CNPase), glial fibrillary acidic protein (GFAP), HLA-DR, Ki67, neuronal nuclei (NeuN), p25α-antigen, and S100β as candidates for future cell counting purposes, because these markers visualized specific neuronal and glial cell bodies. However, significant negative correlation between staining result and formalin fixation was observed by blinded scoring of staining for CD45/LCA, CNPase, GFAP, and NeuN in brain specimens fixed by immersion and stored up to 10 years in 4% formalin solution at room temperature, independent of donor sex and postmortem interval. In contrast, improved preservation of NeuN and CNPase staining, and full preservation of GFAP and CD45/LCA staining in tissue fixed by perfusion and stored for up to 3 years in 0.1% paraformaldehyde solution at 4C, indicated that immunohistochemistry can be performed in well-preserved biobank material.
S
Large collections of human brain material in universities and hospital research departments are available for neurodevelopmental and neuropathological studies (Kostovic et al. 1991; Schmitt et al. 2007; see also www.brainnet-europe.org and www.intbbn.org). To preserve tissue for future analyses, brain banks often store the tissue in fixative, primarily buffered formaldehyde solutions—in some cases for decades (Evers and Uylings 1994, 1997; Perl et al. 2000; Pelvig et al. 2003; Pelvig et al. in press; Samuelsen et al. 2003; Schmitt et al. 2007). It is well known that prolonged fixation with formaldehyde leads to covalent alterations and cross-binding of the tissue molecules, giving rise to structural and electrostatic changes of the antigens (Puchtler and Meloan 1985; Mason and O'Leary 1991; Boenisch 2002, 2005). Masking of antigens can to some extent be reversed using epitope retrieval techniques. Heat-induced epitope retrieval (HIER), in which the tissue section is heated in a buffer (Shi et al. 1991; Pileri et al. 1997; Evers and Uylings 2000; Boenisch 2001; Kahveci et al. 2003), and treatment with proteolytic enzymes (PrER), which leads to degradation of cross-bound protein in the tissue (Pileri et al. 1997; Frost et al. 2000; Kahveci et al. 2003), are the most widely used epitope retrieval techniques and have been successfully applied to a broad range of antibodies and used in standardized protocols for research and diagnostic purposes (www.nordiqc.org and www.ukneqasicc.ucl.ac.uk).
This study was initiated to identify antibodies and protocols that could visualize neurons and glia in formalin-fixed human brain sufficient to meet the requirements for quantitative stereological studies and to investigate the effect of prolonged fixation on staining quality. In the first part of the study, we used 29 different antibodies, directed against the major types of brain cells and immature neurons and glia, in combination with different epitope retrieval methods and different detection systems to stain tissue arrays containing human brain specimens that had been fixed for 24 hr and up to 10 years. On this basis, we identified protocols for candidate antibodies for future stereological applications, visualizing cell bodies of target cells. In the second part of the study, we applied staining protocols for detection of four candidate antibody combinations—neuronal nuclei (NeuN), 2′, 3′ cyclic nucleotide phosphatase (CNPase), glial fibrillary acidic protein (GFAP), and leukocyte common antigen (CD45/LCA)—on a tissue array of parietal cortex from human brains fixed from 8 days to 10 years. The staining quality was scored, and the data were analyzed for correlation between the staining result and fixation, using stratification for sex and postmortem interval (PMI).
Human brain material in tissue array A
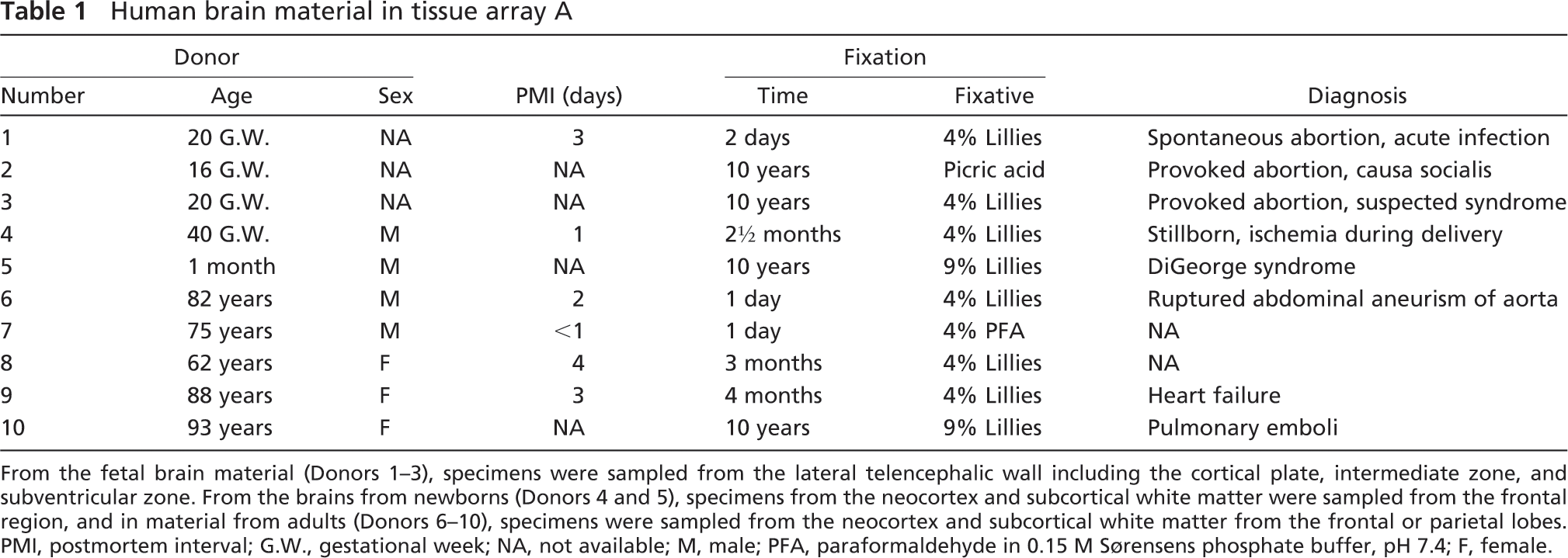
From the fetal brain material (Donors 1–3), specimens were sampled from the lateral telencephalic wall including the cortical plate, intermediate zone, and subventricular zone. From the brains from newborns (Donors 4 and 5), specimens from the neocortex and subcortical white matter were sampled from the frontal region, and in material from adults (Donors 6–10), specimens were sampled from the neocortex and subcortical white matter from the frontal or parietal lobes. PMI, postmortem interval; G.W., gestational week; NA, not available; M, male; PFA, paraformaldehyde in 0.15 M Sørensens phosphate buffer, pH 7.4; F, female.
Materials and Methods
Human Tissue
The use of archival human brain tissue for this study was approved by the Danish Biomedical Research Ethical Committee for the Region of Southern Denmark (permission number S-20070065). All brains included in the study had been subjected to routine neuropathological examination, and no neuropathological changes had been observed.
For screening of antibodies, two tissue arrays were made from identical samples (tissue array A; Table 1): one as paraffin embedded material and one as frozen material. Both contained 16 specimens from the neocortex, white matter, or pons from normal adult, perinatal, and fetal brains. Samples from Donors 2, 3, 5, 8, 9, and 10 were kindly provided by Dr. Bente Pakkenberg, The Research Laboratory for Stereology and Neuroscience, Bispebjerg Hospital, Copenhagen, Denmark (Danish Biomedical Research Ethical Committee for the Region of Sealand, permission number KF 01-068/98 and KF 01-328/98). Samples from Donors 1, 4, 6, and 7 were from the biobank at the Department of Pathology, Odense University Hospital (OUH), Odense, Denmark.
To analyze the effect of fixation time on the staining result, tissue array B was composed of paraffin-embedded material from specimens sampled in the postcentral gyrus of the neocortex from 29 human brains fixed by immersion, ranging from 8 days to 10 years (Table 2). The samples from Donors 1 and 8–29 were from the biobank at the Department of Pathology, OUH. Samples from Donors 2–7 were from brains donated to the Department of Anatomy and Neurobiology, The Institute of Medical Biology, University of Southern Denmark. The material from Donors 2–6 was fixed by perfusion of the head with 4% Lillies phosphate-buffered formalin solution (PBFS), pH 7.0 (Lillies PBFS: 4% w/v formaldehyde, 75 mM phosphate buffer; Sygehusapotek Fyn, OUH) through the internal carotid artery. The brain was removed from the skull and immersed in 4% paraformaldehyde (PFA) in 0.15 M Sørensens phosphate buffer, pH 7.4, and fixed for 2 weeks at 4C before routine pathology. For long-term storage, this material was kept in 0.1% PFA in 0.15 M Sørensens phosphate buffer, pH 7.4, at 4C. The brain from Donor 7 was fixed by immersion only.
Paraffin sections were prepared as 5-μm-thick serial sections mounted on 75-μm capillary gap slides (S2024; Dako Cytomation, Dako Nordic a/s, Glostrup, Denmark). Sections were stored in sealed boxes at 4C. Sections from tissue array A were used within 6 months after sectioning, and sections from tissue array B were used within 2 weeks. Cryostate sections were prepared as 10-μm-thick serial sections, mounted on 75-μm capillary gap slides, and stored in sealed boxes containing silica gel at −20C and used within 1 year after sectioning.
Human brain material in tissue array B
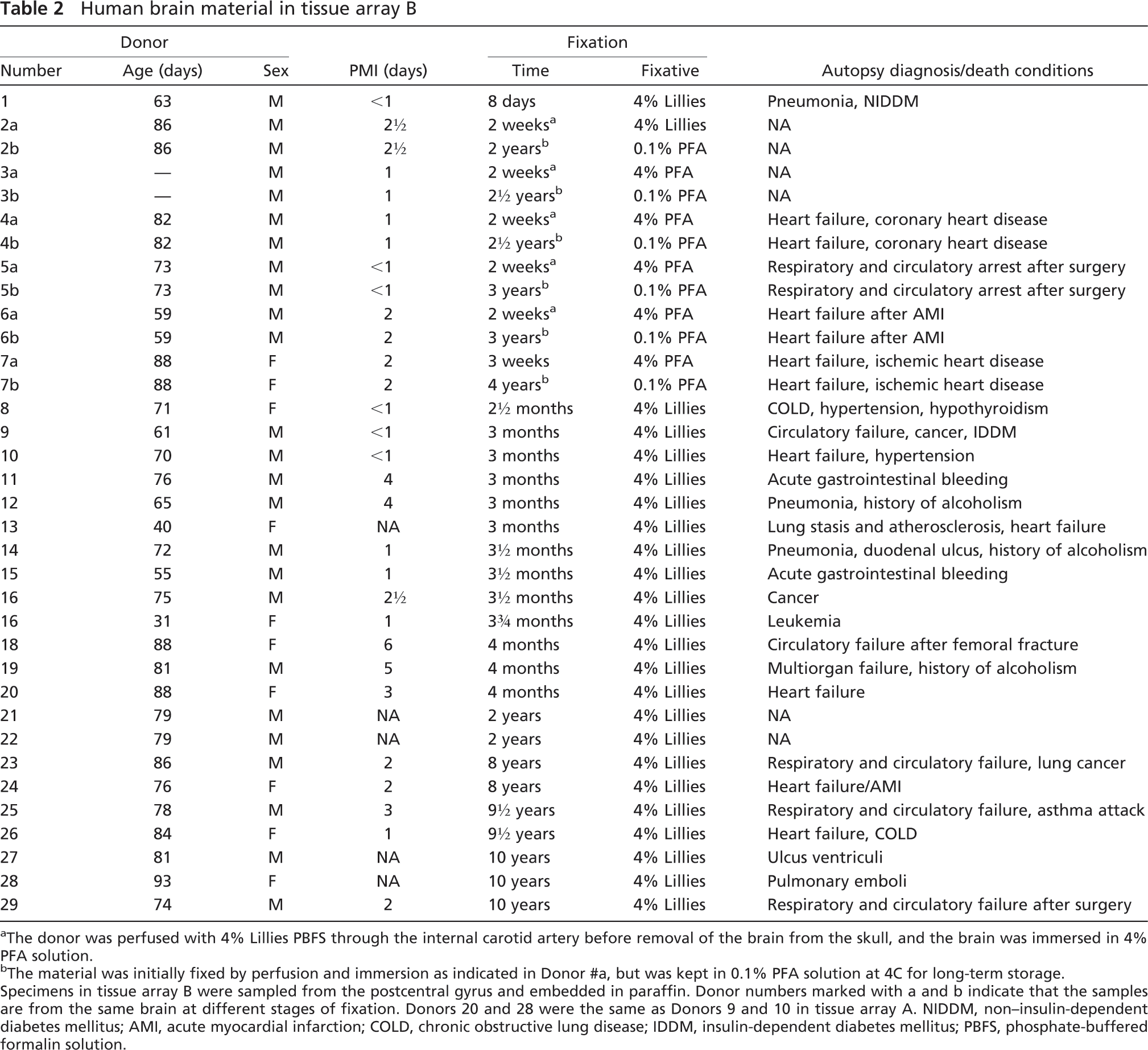
a The donor was perfused with 4% Lillies PBFS through the internal carotid artery before removal of the brain from the skull, and the brain was immersed in 4% PFA solution.
b The material was initially fixed by perfusion and immersion as indicated in Donor #a, but was kept in 0.1% PFA solution at 4C for long-term storage. Specimens in tissue array B were sampled from the postcentral gyrus and embedded in paraffin. Donor numbers marked with a and b indicate that the samples are from the same brain at different stages of fixation. Donors 20 and 28 were the same as Donors 9 and 10 in tissue array A. NIDDM, non–insulin-dependent diabetes mellitus; AMI, acute myocardial infarction; COLD, chronic obstructive lung disease; IDDM, insulin-dependent diabetes mellitus; PBFS, phosphate-buffered formalin solution.
Immunohistochemistry
Paraffin sections were heated for 45 min at 60C to improve adhesion of the section to the slide and deparaffinized in xylene for 4, 3, and 3 min, followed by rehydration in ethanol concentrations decreasing from 99% ethanol to 70% ethanol by several steps lasting 1 min, ending in running tap water. Sections were rinsed in two shifts of distilled water before epitope retrieval, and afterward, the sections were placed in TNT buffer (0.025 M Tris, pH 7.5, 0.03 M NaCl, and 0.05% Tween-20 in distilled water) for staining.
Cryostate sections were used to test antibodies when it was suspected that the antigens could be sensitive to organic solvents. Cryostat sections were washed in several shifts of TNT buffer, and no epitope retrieval was performed, because boiling made sections detach from the glass slides.
HIER was conducted using a microwave oven (Panasonic NN-K655; Skousen, Odense, Denmark) by heating the sections immersed in buffer in plastic Hellendahl vials (ProHosp Denmark a/s; Værløse, Denmark) for 9 min at 880 W and 15 min at 440 W, leaving the sections for 15 min in the gradually cooling buffer. Alternatively, a thermostat-regulated pressure cooker (Digital Decloaking Chamber; Biocare Medical, purchased at ProHosp) was used using the preset program: (1) initial heating and pressurizing to 125C, (2) step SP1 at 125C for 30 sec, (3) step SP2 at 90C for 10 sec, and (4) depressurizing of the pressure cooker. The lid was opened, and the sections were left in the buffer within the boiling chamber for 15 min to cool down. Four different buffers were tested: Tris-EGTA-buffer (TEG; 10 mM Tris, 0.5 mM EGTA, pH 9.0), citrate buffer (C; 10 mM citrate, pH 6.0) (both purchased from Sygehusapotek Fyn, OUH), Target Retrieval Solution diluted 1 + 9 in distilled water (TRS; S1699, Dako Cytomation), and Borg Decloak epitope retrieval buffer (Borg; Biocare Medical, purchased at ProHosp). For PrER, sections were rinsed 2 × 2 min in distilled water and 2 × 2 min in TBS at 37C. Sections were treated with pepsin solution (0.004 g/ml pepsin in 0.01 M HCl; P-7012, Sigma-Aldrich Denmark a/s, Vallensbæk Strand, Denmark) for 20 min or protease solution (0.0005 g/ml Protease type XIV in TBS; P-5147, Sigma) for 15 min at 37C, followed by rinsing in several shifts of TBS at room temperature. For some antibodies, a combination of HIER and PrER was tested.
Staining was performed in a Dako Techmate-500 capillary gap staining-instrument (Dako Cytomation) using the buffers and reagents of the Dako ChemMate system (K5001 and K5006; Dako Cytomation). Three detection systems were applied according to the vendors recommendations: Vectastain Universal Elite ABC Kit (PK-6200; Vector Laboratories, purchased from VWR, Rødovre, Denmark); Dako ChemMate LSAB system, universal mouse/rat/rabbit (K5001), and Dako Envision+ peroxidase-labeled polymer (K4061; both Dako Cytomation). The sequence of the staining program was adjusted to fit the staining procedure recommended by the manufacturer of the detection system. For experiments using Dako ChemMate LSAB system and Vectastain Universal Elite ABC, the staining sequence was (1) TNT buffer, (2) blocking of endogenous biotin (Bussolati et al. 1997) using an avidin/biotin blocking kit (SP-2001; Vector Laboratories) diluted 1 + 5 in TBS, (3) TNT buffer, (4) 30-min incubation in the primary antibodies diluted in Dako ChemMate antibody diluent (S2022; Dako Cytomation), (5) TNT buffer, (6) 30-min incubation in biotinylated secondary antibody solution, (7) TNT buffer, (8) blocking of endogenous peroxidase using the Dako ChemMate HP-Block solution (S2023; Dako Cytomation), (9) TNT buffer, (10) 25-min incubation in horseradish peroxidase (HRP)-labeled streptavidin solution, (11) TNT buffer, (12) chromogenic reaction for 3 × 5 min in Dako DAB+-chromogenic kit, and (13) TNT buffer. The program sequence for staining with Dako Envision+ peroxidase-labeled polymer (K4061; both Dako Cytomation) was modified by removing Step 2 blocking of endogenous biotin and Step 10 incubation with HRP-labeled streptavidin solution. After immunohistochemistry, sections were counterstained in Mayer acidic hemalum for 10 min, rinsed for 10 min in running tap water, and coverslipped with Aquatex (1.08562.0050; Merck, purchased at VWR International a/s, Albertslund, Denmark).
Primary antibodies
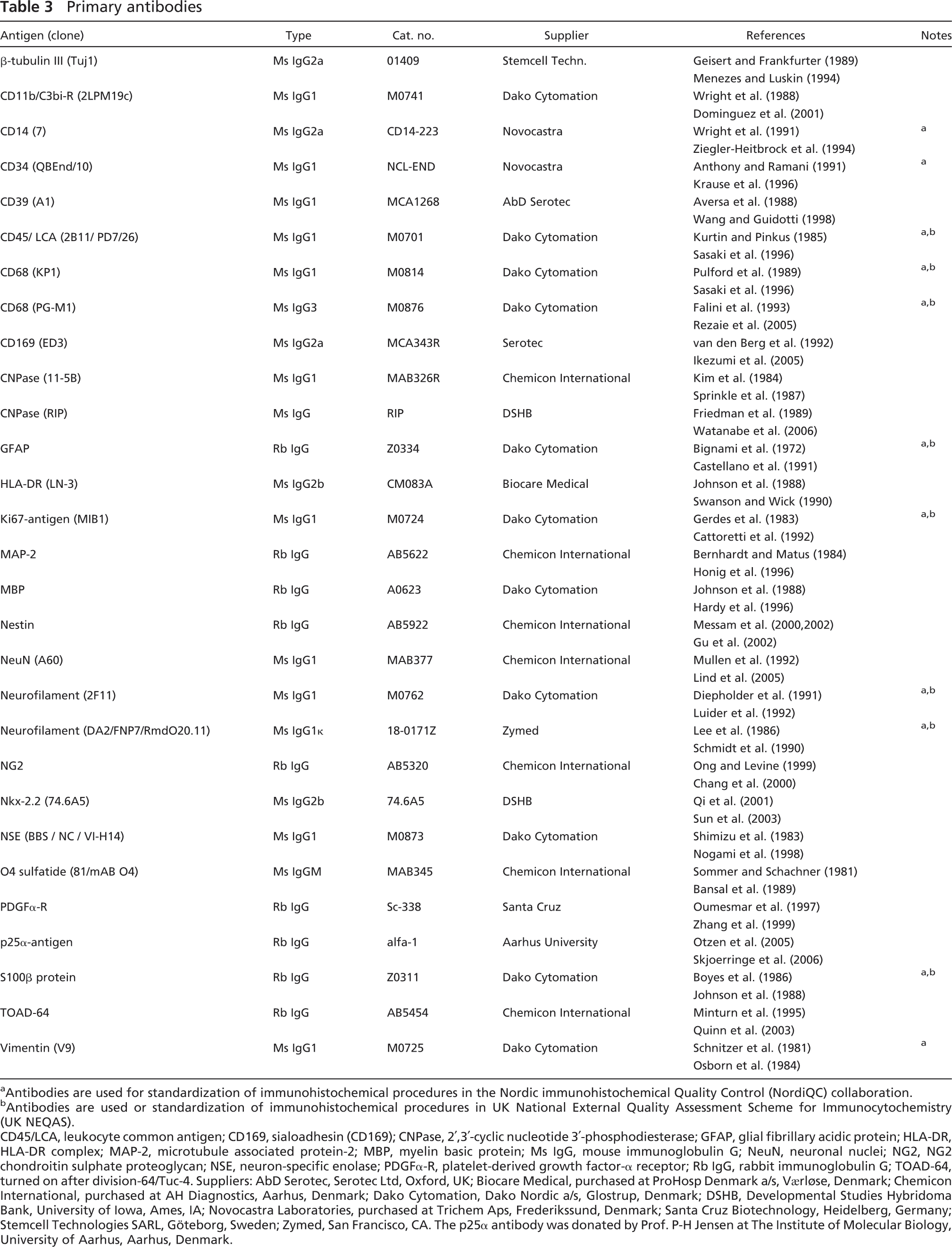
a Antibodies are used for standardization of immunohistochemical procedures in the Nordic immunohistochemical Quality Control (NordiQC) collaboration.
b Antibodies are used or standardization of immunohistochemical procedures in UK National External Quality Assessment Scheme for Immunocytochemistry (UK NEQAS).
CD45/LCA, leukocyte common antigen; CD169, sialoadhesin (CD169); CNPase, 2′,3′-cyclic nucleotide 3′-phosphodiesterase; GFAP, glial fibrillary acidic protein; HLA-DR, HLA-DR complex; MAP-2, microtubule associated protein-2; MBP, myelin basic protein; Ms IgG, mouse immunoglobulin G; NeuN, neuronal nuclei; NG2, NG2 chondroitin sulphate proteoglycan; NSE, neuron-specific enolase; PDGFα-R, platelet-derived growth factor-α receptor; Rb IgG, rabbit immunoglobulin G; TOAD-64, turned on after division-64/Tuc-4. Suppliers: AbD Serotec, Serotec Ltd, Oxford, UK; Biocare Medical, purchased at ProHosp Denmark a/s, Værløse, Denmark; Chemicon International, purchased at AH Diagnostics, Aarhus, Denmark; Dako Cytomation, Dako Nordic a/s, Glostrup, Denmark; DSHB, Developmental Studies Hybridoma Bank, University of Iowa, Ames, IA; Novocastra Laboratories, purchased at Trichem Aps, Frederikssund, Denmark; Santa Cruz Biotechnology, Heidelberg, Germany; Stemcell Technologies SARL, Göteborg, Sweden; Zymed, San Francisco, CA. The p25α antibody was donated by Prof. P-H Jensen at The Institute of Molecular Biology, University of Aarhus, Aarhus, Denmark.
Analysis of Staining Results and Statistics
The staining results from the screening of antibodies in tissue array A were scored by two investigators (LL and IDS) on a scale from 0 to 5: 0, no staining; 1, weak specific signal; 2, weak specific signal giving the impression of lack of stained cells or structures or high level of nonspecific staining; 3, acceptable level of specific signal; 4, high level of specific staining considered optimal for cell counting; 5, very high level of staining that, however, compromised the identification of the cellular nucleus. The results of the stainings for NeuN, GFAP, CNPase, and CD45 in tissue array B were scored by two investigators (BF and HDS) blinded to the fixation time and origin of the specimens. For each staining, five sections resulting from three separate experiments were rated on a scale from 0 to 4 (Table 4). The classification was inspired by the recommendations given by NordiQC (www.nordiqc.org) for evaluation of immunohistochemical staining results. For each staining, a mean score, SD, and SEM were calculated. Statistical analysis for correlation between scores and fixation time was done using the Spearman
Scale for scoring of staining results in tissue array B

Photodocumentation
Sections were photographed using an Olympus DP70 digital camera mounted on an Olympus BX51 microscope and connected to a PC with the Olympus DP software (Olympus Danmark a/s, Ballerup, Denmark). Adobe Photoshop CS was used to adjust contrast of and to set up figures.
Results
Identification of Immunohistochemical Markers for Visualization of Neurons and Glia
Screening of candidate markers for human brains cells in tissue array A
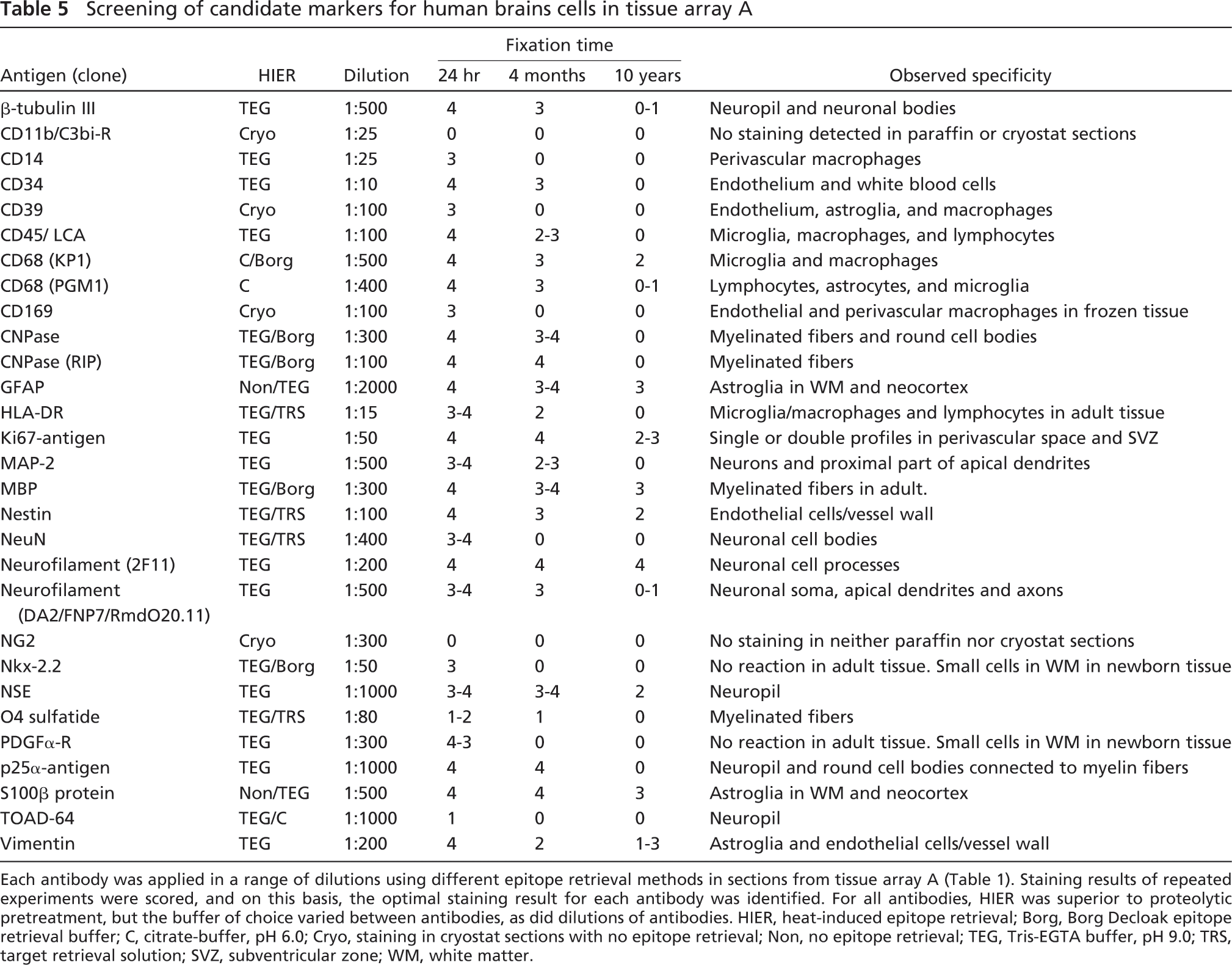
Each antibody was applied in a range of dilutions using different epitope retrieval methods in sections from tissue array A (Table 1). Staining results of repeated experiments were scored, and on this basis, the optimal staining result for each antibody was identified. For all antibodies, HIER was superior to proteolytic pretreatment, but the buffer of choice varied between antibodies, as did dilutions of antibodies. HIER, heat-induced epitope retrieval; Borg, Borg Decloak epitope retrieval buffer; C, citrate-buffer, pH 6.0; Cryo, staining in cryostat sections with no epitope retrieval; Non, no epitope retrieval; TEG, Tris-EGTA buffer, pH 9.0; TRS, target retrieval solution; SVZ, subventricular zone; WM, white matter.
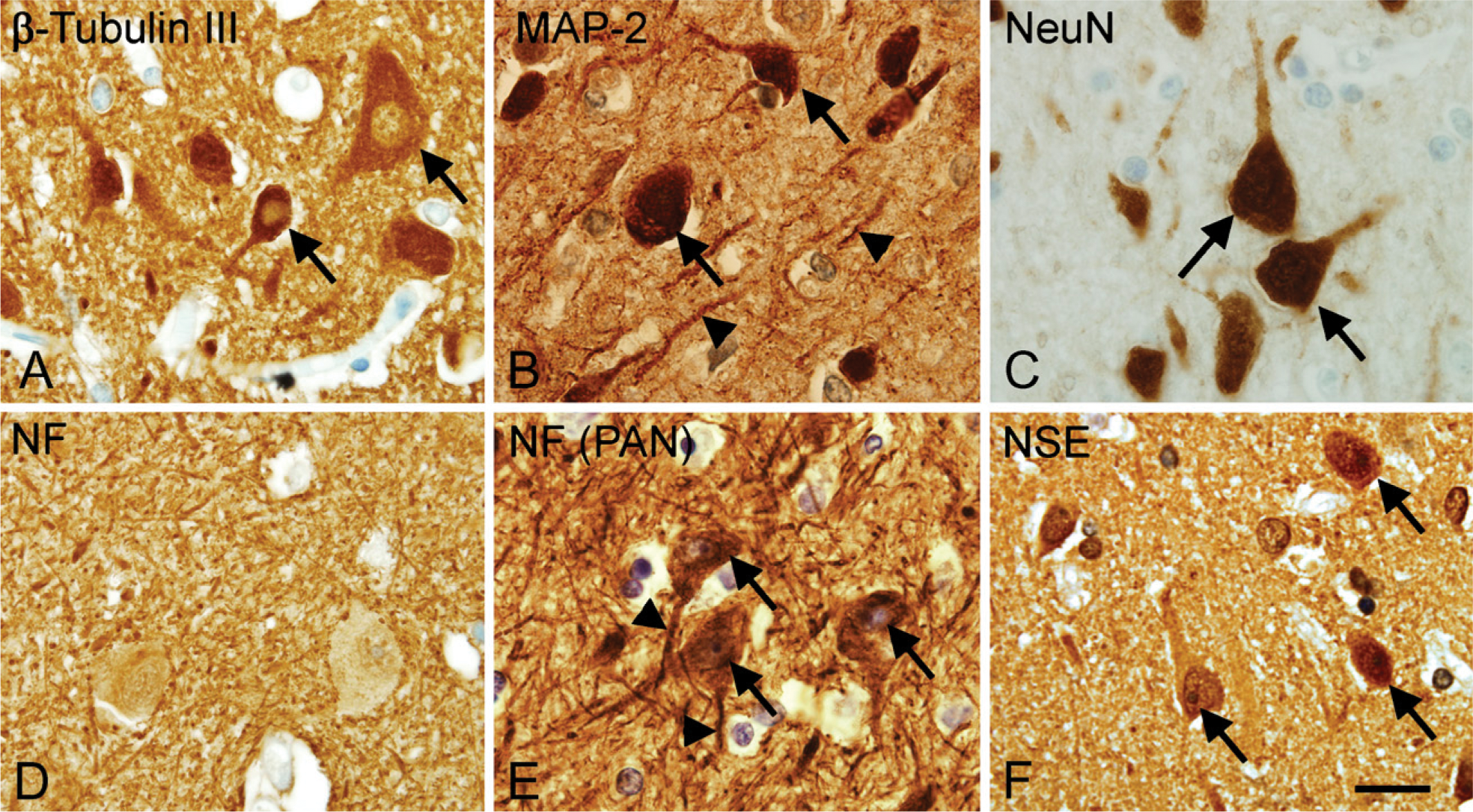
Visualization of neuronal cell bodies and cell processes vary with the choice of antigen/antibody combination. High-magnification micrographs showing the staining for β-tubulin III (
Effect of Epitope Retrieval and Choice of Detection System
In the initial screening, checkerboard titration was performed for each of the antibodies. The results were evaluated qualitatively selecting the combination of epitope retrieval and antibody concentration, resulting in a high level of specific signal and low level of background staining. In our hands, HIER yielded better results than PrER, even in antibodies in which the manufacturer suggested the use of proteolytic pretreatment such as MBP, GFAP, CD68(KP1), CD68(PGM1), and Ki67 (shown for GFAP and MBP in Figure 4). This observation was in agreement with results in recent test reports on www.ukneqasicc.ucl.ac.uk. No difference was observed using the microwave oven or the pressure cooker as a heating source (data not shown), whereas the choice of epitope retrieval buffer (TEG, TRS, Borg, or citrate buffer) greatly influenced the resultant staining depending on the specific antibody (Table 5). The antibodies against CD39 and CD169 gave rise to specific signal in cryostat sections only.
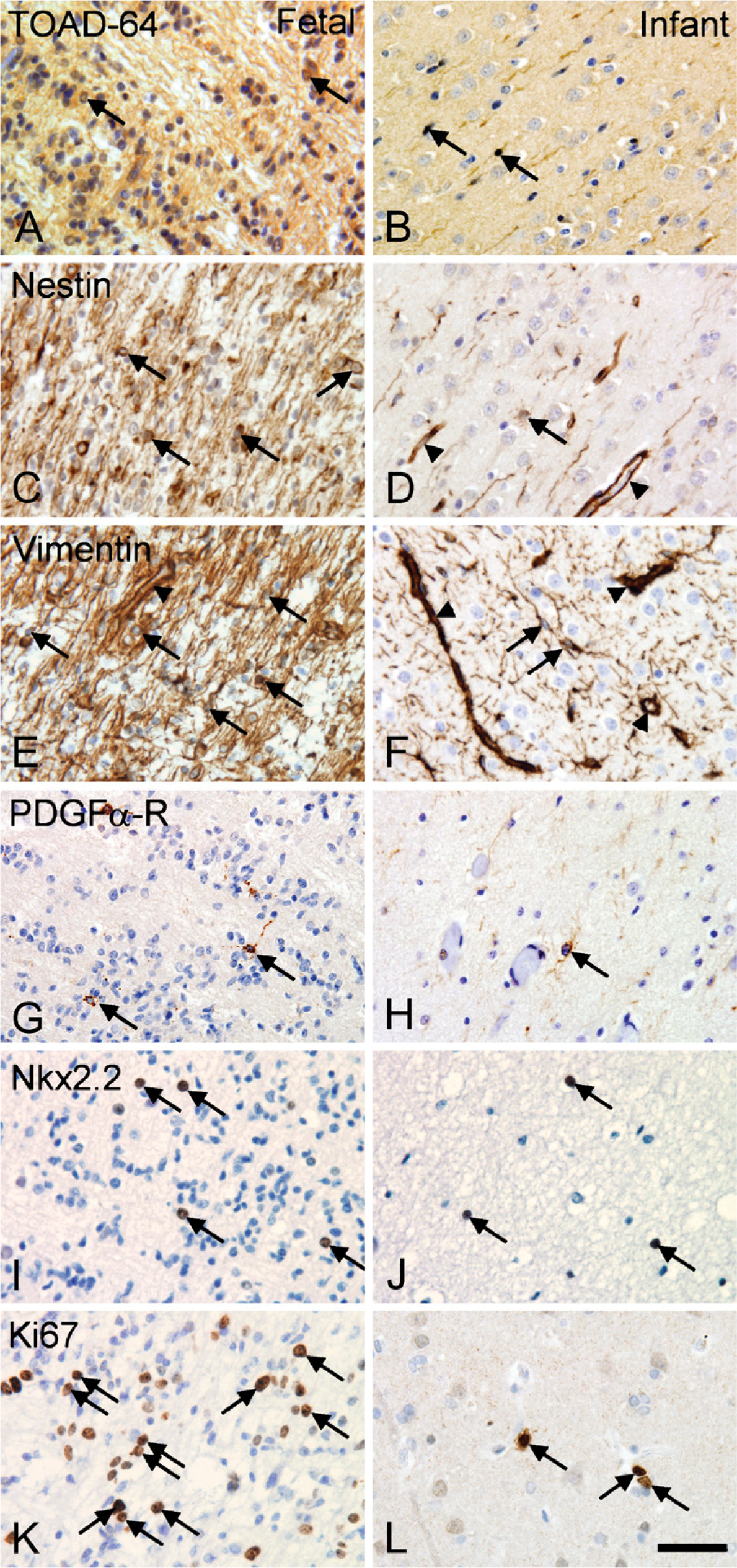
Differences in cellular expression of developmentally regulated antigens in fetal and infant tissue. (
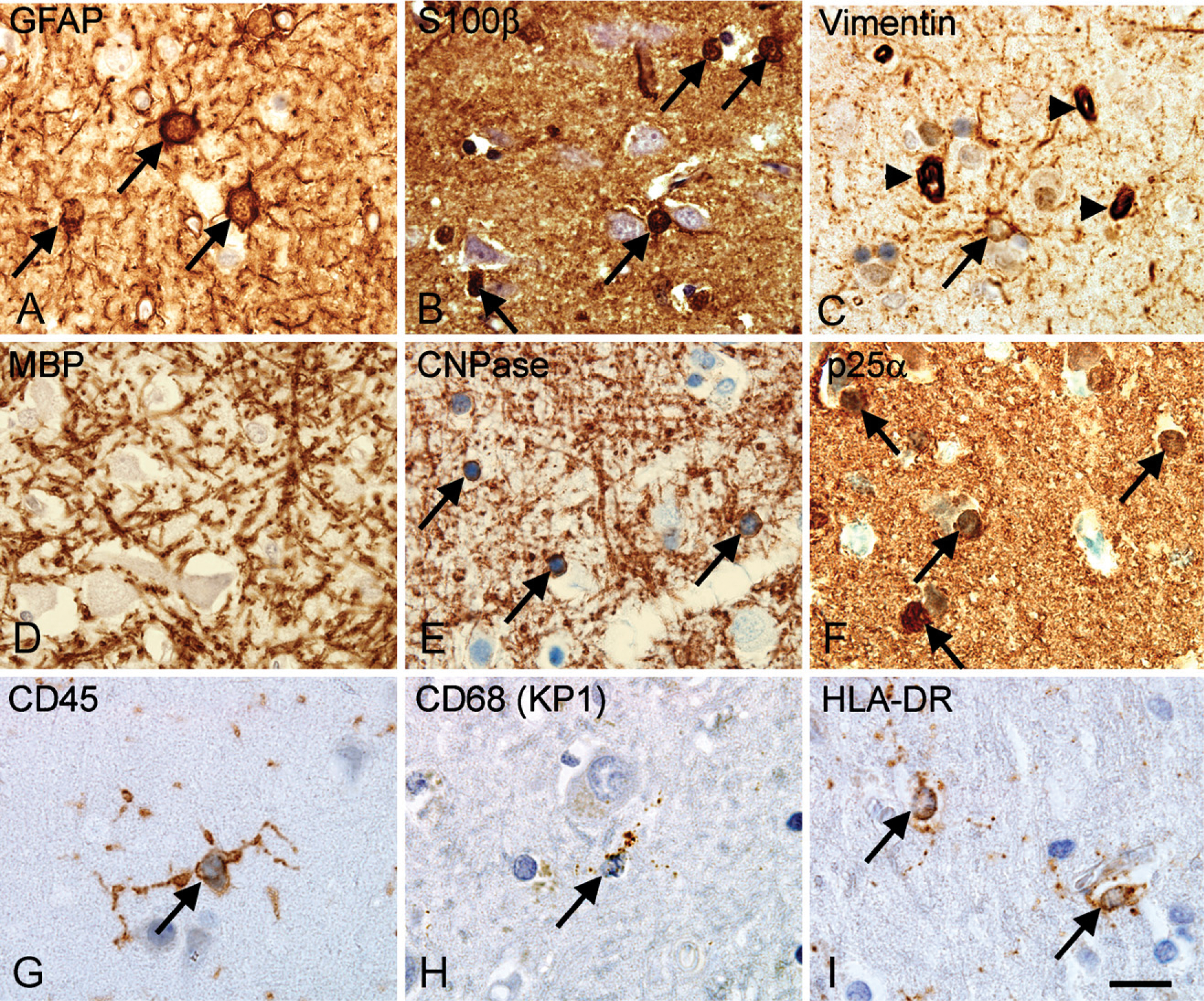
Visualization of astroglial, oligodendroglial, and microglial cell bodies in the parietal neocortex from an adult donor. (
The performance of three detection systems for staining of short- and long-term fixed material was evaluated qualitatively. The Dako ChemMate LSAB-kit gave rise to a high signal level, with considerable background staining in tissue fixed for 24 hr (Figure 5A). The background staining did not compromise results in tissue stored in 4% Lillies PBFS, but the signal level was lower than that of both the Envision+ peroxidase-labeled polymer detection system and the Vectastain Universal Elite ABC kit (Figures 5D–5F). The Envision+ peroxidase-labeled polymer detection system resulted in very high signal levels, visualizing even fine astroglial processes both in tissue fixed in 4% Lillies PBFS for 24 hr (Figure 5B) and 10 years (Figure 5E). The Vectastain Universal Elite ABC kit produced slightly weaker specific signal, yet very low background staining both in short- and long-term fixed specimens (Figures 5C and 5F). Finally, to unveil nonspecific staining caused by the secondary antibodies or detection reagents, sections incubated without the primary antibody or with an inert isotype-specific antibody were included in each staining experiment. With few exceptions, these sections presented no signal, but when applying HIER and the Dako ChemMate LSAB system for staining of samples from adults fixed for 24 hr, a weak staining of white matter, neuropil, and pyramidal neurons was observed (Donors 6 and 7 in tissue array A; data not shown).
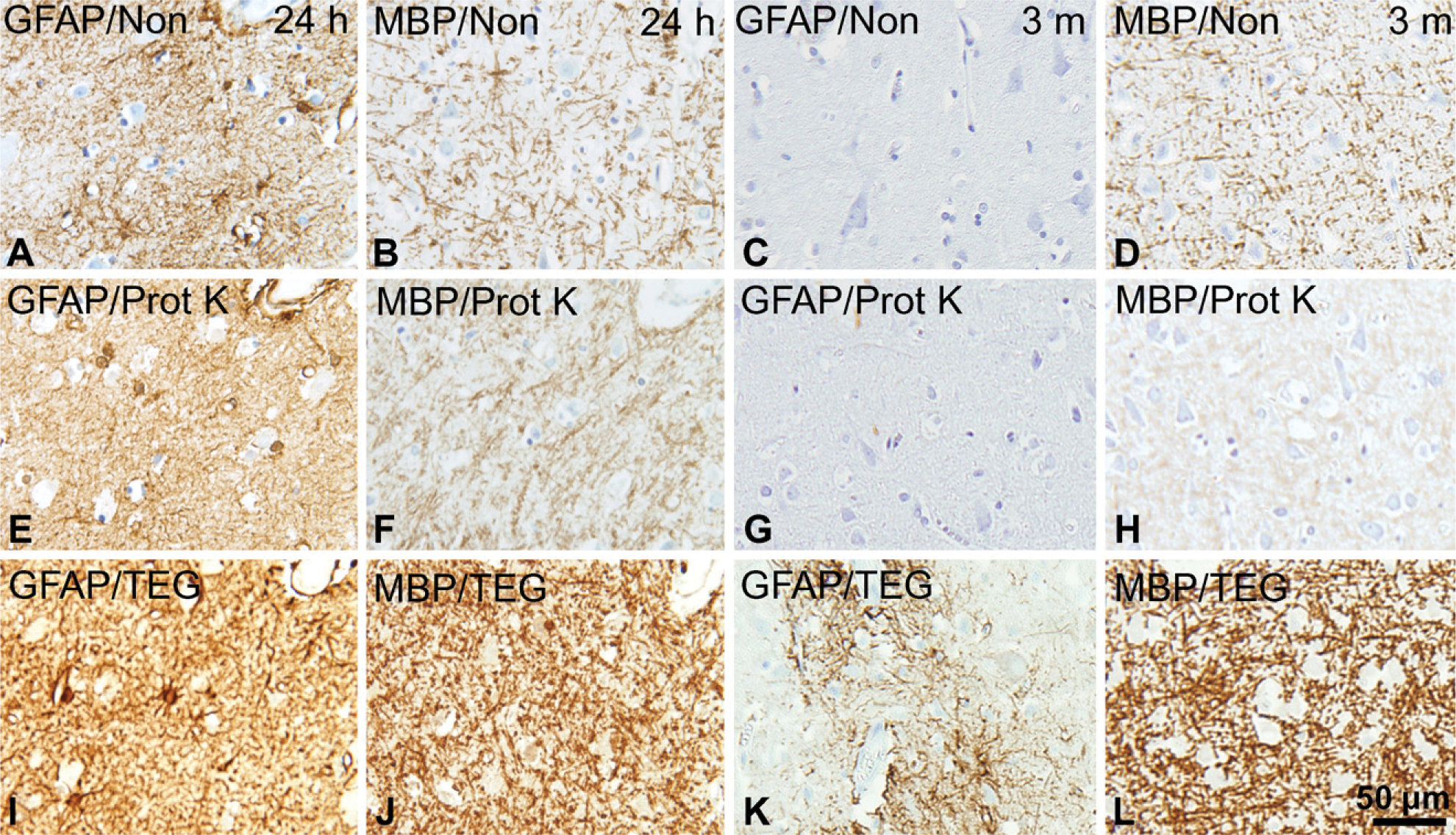
Influence of epitope retrieval technique on staining quality. The effect of HIER and proteolytic epitope retrieval (PrER) were tested on stainings for GFAP and MBP in paraffin sections of adult neocortical tissue fixed for 24 hr and 3 months. (
Effect of Fixation Method and Tissue Storage for Visualization of Specific Antigens
During the initial screening, we identified GFAP, NeuN, CNPase, and CD45/LCA as candidate markers for visualization of the cell bodies of the four major classes of brain cells, noting that the staining for GFAP and CD45/LCA appeared to be relatively robust, whereas NeuN and CNPase were sensitive to formalin fixation. We applied these antibodies in a protocol involving HIER in TEG buffer and detection with the Envision+ peroxidase-labeled polymer detection system to paraffin sections from tissue array B (Table 2). The staining quality of individual samples was scored on a scale from 0 to 4 (Table 4; Figure 6) from five different slides and by two independent observers blinded to the arrangement of the tissue array. For each sample, the mean score and SEM was calculated, whereafter the data were stratified for fixation method (Figures 7 and 8), sex (♂:
NeuN staining was observed in only two of the specimens fixed by simple immersion in 4% Lillies PBFS (Table 2, Donors 1 and 7a), having mean scores of 1.7 and 2.6, respectively. All specimens fixed by immersion for >2.5 months were scored 0, making stratification for sex and PMI redundant (Figures 7A and 7B). Specimens that had been fixed by perfusion before immersion in 4% PFA for 2 weeks obtained mean scores >2 (two of five specimens: 2–2.4; three of five specimens: 3.8–4; Figure 8A). However, although one specimen maintained a score of 3, most specimens had dropped to a mean score of <1 (four of five specimens: 0–0.6) after 36 months of storage in 0.1% PFA at 4C (Figure 8A).
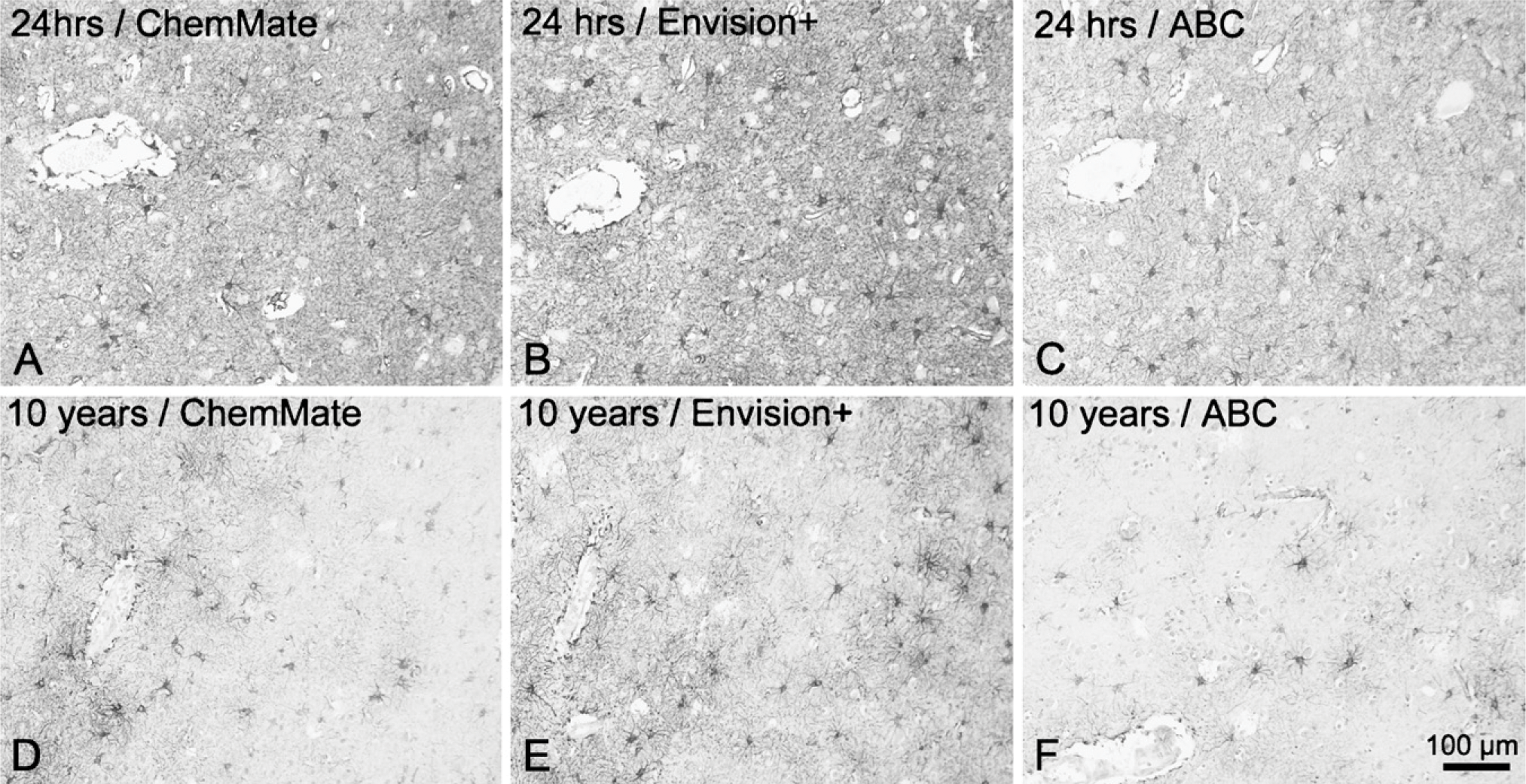
Influence of detection system on staining quality. The visualization of cell bodies and cell processes, illustrated by the visualization of neocortical GFAP+ astrocytes in paraffin sections of adult human brain material fixed for 24 hr (
The mean scores of the CNPase stainings correlated negatively to the storage time in 4% Lillies PBFS at room temperature (Figures 7C and 7D) in both sexes (♂:
In case of GFAP, the mean scores for the GFAP stainings correlated negatively with storage time in 4% Lillies PBFS at room temperature in specimens from male donors (
The mean score of the staining result for CD45/LCA was negatively correlated to the time stored in 4% Lillies PBFS at room temperature (Figures 7E and 7F). As shown for CNPase, this was independent of the sex of the donor (♂:
In the case of GFAP and CD45, the staining sensitivity seemed to be influenced by the cellular activation state. In some samples, astrocytes and microglia showed an activated phenotype with hypertrophic cell bodies and hypertrophic or blunted processes throughout sections, making these cells very easy to identify. In other specimens, microglia expressed very low levels of CD45 and showed a resting phenotype, with thin angulated processes (Figures 5G and 5H). In such samples, there were occasionally small areas corresponding to the territory of one or two microglial cells with no staining, suggesting that CD45 might not be expressed by all resting-like microglia in human brain.
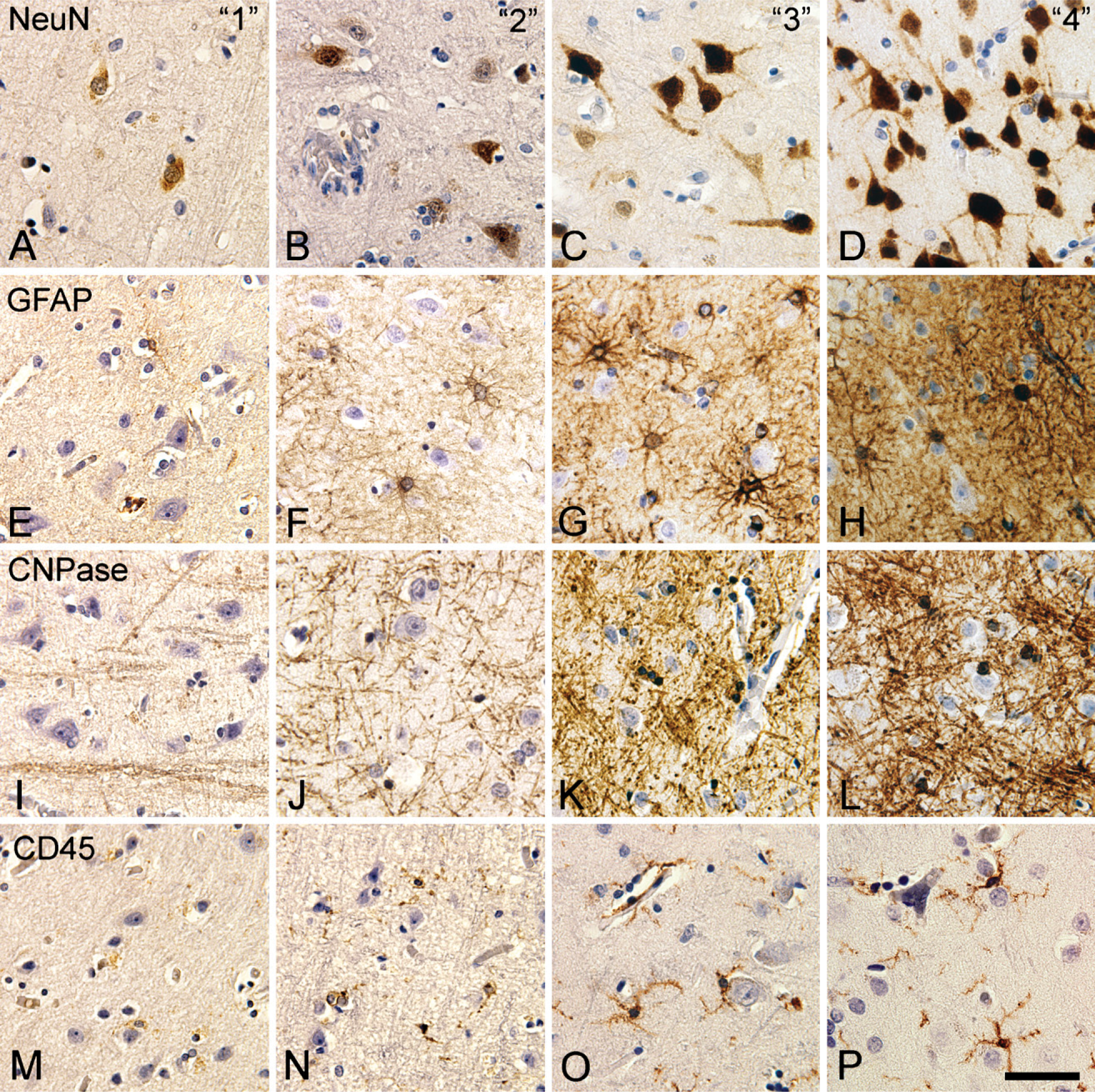
Scoring of immunohistochemical stainings for NeuN, GFAP, CNPase, and CD45. Photomicrographs were acquired in neocortical layer VI in specimens from tissue array B (Table 2) scored as 1 (
Discussion
As a first step in successfully applying stereology and immunohistochemistry to the human brain, the intent of the study was to identify candidate immunohistochemical markers for visualization of the cell bodies of neurons, astrocytes, oligodendrocytes, and microglia, which yield reproducible staining results when applied in human brain tissue obtained from autopsies and stored in formalin fixative solutions.
Even well-designed studies in human postmortem brain tissue based on matched groups will face variations deriving from differences in the quality of the tissue because of differences in biology and conservation of the tissue (Lewis 2002). Several variables influence the conformation of an antigen in a tissue section. Autolysis occurring during the PMI is known to degrade or change tissue antigens, making this an important factor to control for (Lewis 2002; Maleszewski et al. 2007; Schmitt et al. 2007). In this context, the choice of fixation method and fixative solution is of concern, with several reports pointing at perfusion with fixative to yield a more rapid and even fixation, beneficial for immunohistochemistry (Adickes et al. 1997; Lyck et al. 2006; Sharma and Grieve 2006). Finally, the storage time in formaldehyde fixative solutions (Beckstead 1994; Nadji et al. 2005) and the method of embedding or tissue stabilization before sectioning is known to influence the availability of tissue antigens (Werner et al. 1996). The specificity and sensitivity of an immunohistochemical staining method is influenced by the choice of epitope retrieval technique (Pileri et al. 1997; Shi et al. 1997; Frost et al. 2000; Boenisch 2005), by the use of detergents (Weruaga et al. 1998; Heffer-Lauc et al. 2007), or by blocking of disturbing endogenous molecules (Bussolati et al. 1997; Shi et al. 1997). Furthermore, the specificity and avidity of the primary or secondary antibodies can be altered by the conditions of the incubation (Boenisch 1999, 2001, 2002) and by the choice of detection system (Ellis and Halliday 1992; Werner et al. 1996; Pileri et al. 1997).
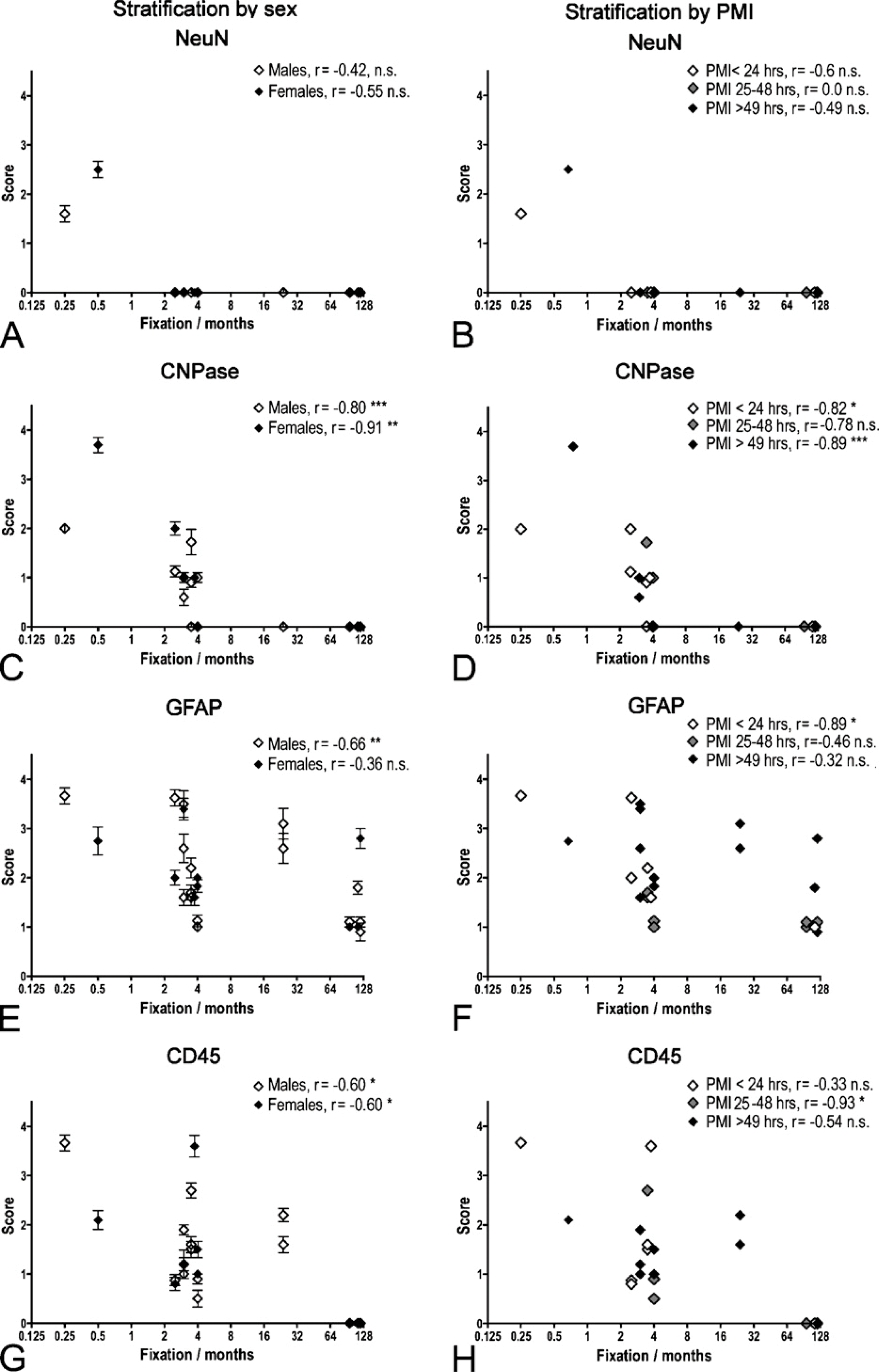
Correlation between immunohistochemical staining result and storage time in 4% Lillies phosphate-buffered formalin solution (PBFS) at room temperature. (
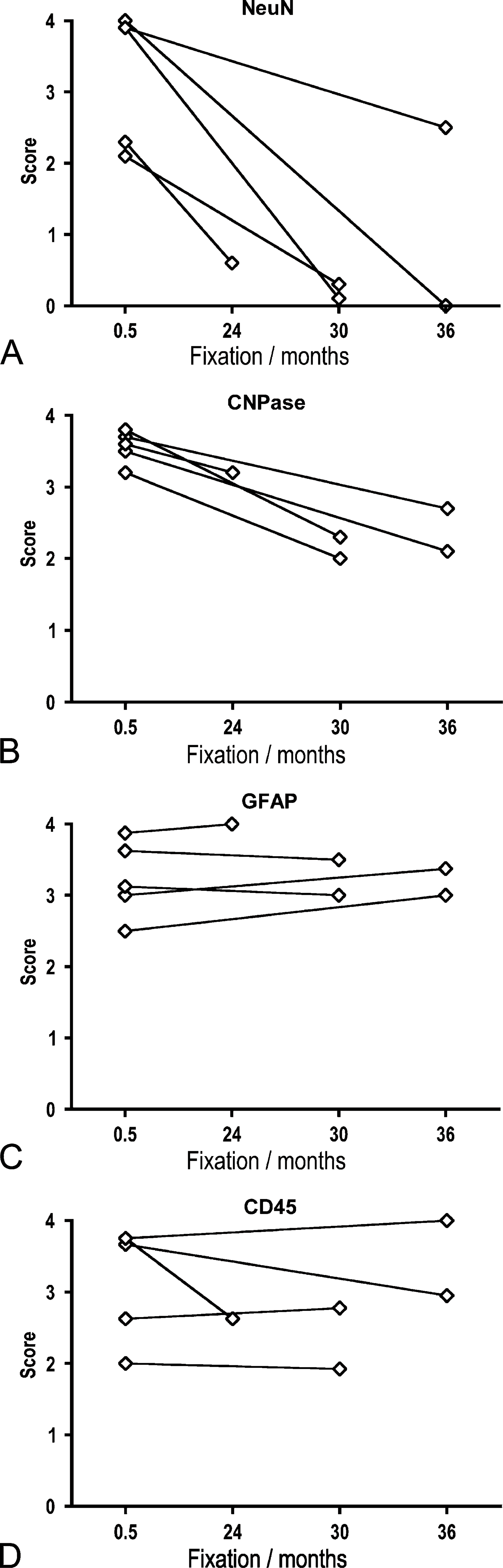
Immunohistochemical staining result in perfusion-fixed brain material. Graphic illustration of changes in immunohistochemical staining results for NeuN (
By checkerboard titration, we systematically tested 29 antibodies in combination with HIER or PrER and three different detection systems in tissue arrays, prepared as either paraffin or cryostat sections. The staining results were then evaluated qualitatively. This strategy has previously been used for establishment and standardization of immunohistochemical protocols for other purposes and in other tissue types (Pileri et al. 1997; Boenisch 2001, 2005; Maleszewski et al. 2007) and in international standardization collaborations (www.nordiqc.com; www.ukneqasicc.ucl.ac.uk). We took advantage of an automated staining procedure. This allowed simultaneous staining of large batches of sections, but required the sections to be maximally 10 μm thick, which is not appropriate for cell counting by either the optical or physical dissector, but suitable for a qualitative evaluation of staining results. Furthermore, the staining protocols, involving 30-min incubation with primary antibodies and secondary reagents at room temperature, were selected on the basis of protocols used in international programs for standardization of immunohistochemical methods (www.nordiqc.com; www.ukneqasicc.ucl.ac.uk).
We tested two PrER procedures and HIER in four different buffers, observing that HIER gave better results than PrER (Figure 4; Table 5). Because we observed no difference in the staining results when conducting HIER using a microwave oven or a thermostat-regulated pressure cooker, we performed HIER in the microwave oven, which was easy to handle with large batches of sections. The result of individual antibodies improved by HIER in different buffers, yet for most of the tested antibodies, HIER with TEG was most effective. Although HIER was efficient in increasing the sensitivity of staining in long-term fixed material, the use of HIER in tissue fixed for 24 hr induced nonspecific staining. Notably, when using the Dako ChemMate LSAB detection system in short-term fixed specimens, we observed weak nonspecific staining of myelinated fibers, neuropil, and pyramidal neurons, similar to nonspecific staining patterns reported earlier due to cross-binding of the secondary antibodies in sections treated with HIER (Ellis and Halliday 1992; Shi et al. 1997; Horobin 1998; Weruaga et al. 1998). This background staining was not present in specimens fixed for 2 weeks or longer, and it was absent in staining performed with the Dako Envision+ peroxidase-labeled polymer detection system or Vectastain Universal Elite ABC kit. Because the Dako Envision+ peroxidase-labeled polymer circumvents the problem with false-positive detection of endogenous biotin after HIER treatment (Bussolati et al. 1997) and gives the highest signal-to-noise ratio, we used this detection system to test the effect of tissue fixation on staining intensity.
Prolongation of the incubation period with the active reagents can to some extent improve the sensitivity of an immunohistochemical staining (Boenisch 2002). This strategy is mainly used to allow for the use of highly dilute antibodies (Heinsen et al. 2000), to improve the signal-to-noise ratio when using polyclonal antibodies and high stringency rinsing (Larsson 1989), or to improve the penetration of staining in thick, free-floating sections (Evers and Uylings 1994, 1997). We believe that the incubation period of 30 min was sufficient to allow binding of primary antibody for the following reasons: (1) titration of the antibody concentration was performed for all antibodies resulting in reproducible results between experiments, (2) tissue array B was designed so that specimens with short and long storage time in fixative were placed next to each other to control for an effect of the arrangement on the array, (3) the scoring was performed by investigators blinded to the arrangement of the specimens in the tissue array, and (4) the use of 30-min incubation during automated staining of sections originally had previously been shown to result in optimal staining through systematic testing in the Department of Pathology, OUH.
Immunohistochemistry in Tissue Stored in Fixative Solution
Most of the literature describing staining with the antibodies tested in this study are based on staining of material fixed in formaldehyde fixatives for hours or a few days, including studies of the expression of NeuN (Sarnat et al. 1998b), vimentin (Sarnat 1998a), MBP (Zecevic et al. 1998), O4, and NG-2 (Back et al. 2001). Even studies testing the effect of HIER on immunostaining focused on specimens fixed up to 1 week (Pileri et al. 1997; Nadji et al. 2005). Only a few studies have addressed the use of immunohistochemistry on tissue kept in formalin for >1 year (Evers and Uylings 1994, 1997). Evers and Uylings (1994, 1997) reported good results for detection of neuronal antigens MAP-2, phosphorylated and non-phosphorylated neurofilaments, calbindin, parvalbumin, calretinin, and neuropeptide Y, but also reported that it was impossible to obtain satisfactory staining results in tissue kept in fixative for ≥8 years. In the initial screening, staining results for most antigens were excellent when applied to specimens from brains stored in fixative for 24 hr (Table 5), yielding results comparable to literature reports. Exceptions to this were the antibodies against CD11b, NG-2, and O4 yielding no staining, neither in paraffin nor cryostat sections. The absence of signal could be caused by degradation of the antigens because both NG-2 and O4 have been detected in unfixed, frozen sections of human brain material sampled with only very short postmortem delay (Chang et al. 2000; Back et al. 2001). In this study, most tissue was collected with a longer postmortem delay, raising the possibility that these antigens might be particularly sensitive to postmortem autolysis. With longer time of fixation, staining results varied, raising a demand for a more rigorous test of the effect of fixation time on the staining result than could be achieved with the suboptimally matched tissue samples in tissue array A (Table 1).
Based on the initial screenings, we tested the effect of fixation time on NeuN, CNPase, GFAP, and CD45, applying the HIER/TEG protocol for epitope retrieval and the Dako Envision+ peroxidase-labeled polymer detection system to tissue array B, which was composed of 29 different specimens sampled in the postcentral gyrus from adult donor brains fixed for 8 days to 10 years. Although perfect matching of the donors was not possible, all donors were selected on the basis of their medical history and diagnoses, avoiding inclusion of donors with conditions that could severely impact the brain. Most of the donor material was fixed by simple immersion of the brain in 4% Lillies PBFS at room temperature, as is the routine at the Department of Pathology, OUH, and at the Brain Bank at Bispebjerg Hospital (Pakkenberg and Gundersen 1997; Samuelsen et al. 2003). In addition, we included five sets of specimens sampled from donor brains that had been fixed by perfusion before immersion-fixation for 2 weeks in 4% PFA and storage in 0.1% PFA solution at 4C for 2–3 years. For both types of fixation, the staining results were excellent when brains had been fixed for 2–3 weeks, yet the results pointed at better preservation of sensitive antigens like NeuN and CNPase in perfusion-fixed specimens. NeuN was very sensitive to fixation, with complete disappearance of immunohistochemical signal after 2–3 months of storage in 4% Lillies PBFS, and the staining signal also deteriorated with storage time in 0.1% PFA at 4C. For GFAP and CD45, excellent staining was obtained in several specimens from immersion-fixed brains stored in 4% Lillies PBFS for up to 4 months, yet preservation of these antigens was improved in perfusion-fixed material stored in 0.1% PFA at 4C, which was also pronounced for CNPase. This difference could be caused by several things. First, the perfusion with fixative resulted in a more even and quick fixation of the brain tissue, preventing degradation of tissue antigens. This observation is in line with recent literature suggesting the use of perfusion fixation for human brains to improve the neuropathological examination caused by better preservation of morphology and tissue antigens (Adickes et al. 1997; Sharma and Grieve 2006; Schmitt et al. 2007). Second, it might be better to store the already fixed tissue at 4C in a solution containing less formaldehyde than the usual conduct of storage. The recent consensus report from The Consortium of Brainnet Europe II set a focus on the importance the preparation, fixation, and storage of brain tissue collected by brain banks to better preserve the tissue molecules (Schmitt et al. 2007), and our data add to the discussion on how to fix and store brain tissue.
Selection of Candidate Markers for Future Cell Counting Studies
The histological material used in this study did not conform to the demands set up for unbiased stereology. First, the 5-μm paraffin sections were not thin enough to qualify for counting of neurons or glial cells using the physical disector (Sterio 1984). For this purpose, sets of serial sections cut at 1–2 μm should be used. Second, the 10-μm-thick cryostat sections did not qualify as thick sections for cell counting by the optical disector (Gundersen 1986, West et al. 1991). To use this tool for counting of large cells like neurons, the final section thickness should be at least 25 μm (Andersen and Gundersen 1999; Dorph-Petersen et al. 2001), and penetration of the staining through the section thickness should be validated by analysis of the z-axis distribution of labeled cells (Dorph-Petersen et al. 2001; Gardella et al. 2003). Recent studies in mice have combined the use of immunohistochemistry and stereology for estimation of total numbers of NeuN+ neurons (Lyck et al. 2007), CD11b+ microglia (Wirenfeldt et al. 2003, 2007), and GABAergic neurons (Muller et al. 2001; Lifshitz et al. 2007) in specific brain regions by using the optical fractionator.
Based on our results, we suggest NeuN for identification of neurons in the human neocortex, because β-tubulin III–, MAP-2–, NF(PAN)-, and NSE-labeled epitopes are also present in neuronal cell processes. GFAP and S100β can be used to label astroglia, although both markers might be suboptimal. GFAP labeling is low in the protoplasmic astrocytes in the neocortex (Korzhevskii et al. 2005) and high in activated astrocytes (Schmidt-Kastner and Szymas 1990, Jessen 2006). It was recently shown that the binding of the polyclonal rabbit anti-GFAP antibody from Dako Cytomation is sensitive to phosphorylation (Tramontina et al. 2007). S100β has been reported to also label subpopulations of oligodendroglial cells (Rickmann and Wolff 1995; Tiu et al. 2000a, b). Only CNPase and p25α identified the cell bodies of oligodendroglia in adult brain. Nkx2.2+ and PDGFα-R+ oligodendroglial precursor cells were observed in only fetal and newborn brain. Because the anti-p25α-antibody was characterized as a marker for oligodendrocytes just recently (Otzen et al. 2005; Skjoerringe et al. 2006), more knowledge on its specificity is needed before applying this marker in quantitative studies, rendering CNPase the candidate for identification of oligodendrocytes. In case of microglia, staining for CD45/LCA provided better visualization of the cell body than staining for CD68(KP1) and labeled much more microglia than staining for HLA-DR. Microglial expression of hematogenous cell markers is known to depend on the state of activation (Mittelbronn et al. 2001; Perry 2003; Raivich and Banati 2004; Ladeby et al. 2005). We observed some variation in intensity of staining and morphology of microglia, with appearance of coarse, intensely stained profiles in samples from some brains. This raises the question if perimortal activation of microglia may occur, influencing the detection of these cells. Finally, monocytes and perivascular cells also express these markers (Sasaki et al. 1996; Mittelbronn et al. 2001; Ladeby et al. 2005), and the investigator should be able to distinguish between these cells and microglia during quantitative studies. A recent paper suggested the use of an antibody against IBA-1 for labeling of microglia in human brain (Ahmed et al. 2007). This was not tested in this study but could be another potential marker for cell counting purposes.
Although the optical disector has already proven its value for counting of immunohistochemically labeled cells, it may be advantageous to use the physical disector because of transparency of sections with intense specific labeling of the neuropil. The presented staining methodology could easily be implemented in sets of parallel 1- to 2-μm paraffin sections for counting using the physical disector, because the staining method is already optimized for staining of thin sections in large batches. For example, it may be possible to use the protocols for labeling of neurons expressing β-tubulin III, MAP-2, NF(PAN), or NSE in a physical disector design, just as counting of GFAP+ astrocytes should be done using the physical disector because of limited penetration of staining into thick sections (Lyck et al. 2006). Furthermore, in a recent stereological study of microglial responses to axonal lesion, it was noted that reactive microglia were present in the tissue as “clusters,” making it difficult to distinguish individual cells when using the optical disector (Wirenfeldt et al. 2007). Thus, it is necessary to test the immunohistochemical staining in conjunction to actual application to clarify whether the staining is reproducible between subjects and whether it allows identification of the individual cells in the specimen in a way that conforms with the rules for stereological counting.
In conclusion, based on a screening of a range of frequently used antibodies along with epitope retrieval techniques and detection systems, we identified a panel of candidate markers for future stereological studies. We also observed considerable loss of immunohistochemical staining signal in tissue specimens stored for long periods of time in 4% Lillies PBFS at room temperature, which has been widely used in tissue banks. For some markers, this loss could be delayed and possibly reduced in the long term by perfusion fixation before storage in 0.1% PFA at 4C. We believe that the application of immunohistochemistry in future stereological studies of the human brain will be possible with careful selection and validation of staining methods and by the use of well-preserved material.
Footnotes
Acknowledgements
The Augustinus Foundation, The Beckett-Foundation, The Carlsberg Foundation, The Danish Multiple Sclerosis Society, Fonden til Lægevidenskabens Fremme, The Gangsted Family Foundation, the Lundbeck Foundation, The Danish MRC, and The Velux Foundation of 1981 supported this work by grants to L.L. and B.F.
We thank Ole Nielsen, Lisbeth Mortensen, Inger Nissen, and Lene Jørgensen for technical assistance and Dr. Pakkenberg, Research Laboratory for Stereology and Neuroscience, Bispebjerg Hospital, and Dr. Kock, Department of Pathology, Odense University Hospital, for donation of tissue samples. Dr. Fenger, University of Southern Denmark, donated anti-Nkx-2.2 antibody, and the anti-Rip antibody was a gift from Dr. Owens, Montreal Neurological Institute, McGill University, Montreal, Canada.