
Abstract
Lung resistance-related protein (LRP) is an integral part of the multidrug resistance (MDR) phenotype involved in cell resistance toward xenobiotics or chemotherapy. The aim of this study was to compare the intracellular localization and cell expression of LRP in normal bronchial cells and their tumoral counterparts from non-small cell lung cancer (NSCLC). LRP expression was also investigated concurrently with DNA ploidy and chromosome 16 (lrp gene locus) aberrations. Confocal microscopy showed that LRP localization was exclusively intracytoplasmic regardless of the cell type and was never observed in the nuclear pore complex. Flow cytometry demonstrated a similar level of LRP expression in normal bronchial cells and in cancer cells from NSCLC samples. FISH analysis, performed to evaluate the number of chromosome 16 and lrp loci, demonstrated a significant gain of chromosome 16 in DNA aneuploid tumors. Furthermore, we did not find any link between LRP expression and DNA ploidy status or chromosome 16 number. These results suggest that LRP expression observed in NSCLC, maintained through the carcinogenesis process of respiratory cells, is not altered by the increased number of copies of chromosome 16 and probably controlled by mechanisms different from those of MRP1 expression, whereas both proteins are associated with the MDR phenotype.
Keywords
M
LRP, initially described in non-small cell lung cancer (NSCLC) cell lines lacking P-gp170 (Kuiper et al. 1990; Scheper et al. 1993), is a 110-kDa protein encoded by the lrp gene localized on locus 16p11.2, proximal to the mrp1 gene on the short arm of chromosome 16 (Slovak et al. 1995). LRP has been found to be identical to the human major vault protein (MVP), which contributes to 70% of the mass of vault particles (Kedersha and Rome 1986; Scheffer et al. 1995). Vault particles are complex ribonucleoprotein particles that, in addition to the MVP, also contain two minor 193- and 240-kDa vault proteins and a small RNA, vRNA. Vault particles are arranged into hollow barrel flower-like structures measuring ∼35 × 65 nm with a nuclear mass of ∼13 MDa, with 8-fold symmetry, an invaginated waist, and two protruding caps (Kedersha et al. 1991). It has been calculated that each petal contains six copies of LRP and one vRNA molecule, for a total of 96 and 16 molecules, respectively, in each vault particle (Kedersha et al. 1991). Vaults are mainly cytoplasmic organelles, but some authors have reported that a small number of vaults (∼5%) are localized in the nuclear membrane and in the nuclear pore complex (NPC), supporting the hypothesis that vaults could act as shuttles between the nucleus and the cytoplasm (Chugani et al. 1993).
Respiratory airway cells continuously exposed to airborne pollutants, particularly genotoxic xenobiotics such as tobacco smoke, like all epithelial cells localized at the interface with the external environment are known to possess well-developed cell detoxification mechanisms, particularly the MDR phenotype (Dingemans et al. 1996). We, like others, have already explored the localization of two ABC transporters in airway tissues: P-gp170, a product of the mdr1 gene expressed in normal airways at the apical cell membrane of ciliated cells (Lechapt-Zalcman et al. 1997; Wioland et al. 2000), whereas MRP1 encoded by the mrp gene is expressed both at the basolateral cell membrane of ciliated and basal cells and at the intracellular level (Brechot et al. 1998; Wioland et al. 2000). However, few studies have precisely investigated the intracellular localization of LRP in normal respiratory cells (Berger et al. 2000).
Primary lung cancers, which arise from airway epithelial cells, are currently the leading cause of cancer deaths worldwide. They are classified into two main histological subtypes: small cell lung cancer (SCLC) and NSCLC, constituting ∼85% of all lung cancers. Tumor resistance in patients receiving chemotherapy alone or combined with radiotherapy represents a major problem in treatment of NSCLC (Rosell et al. 2002). As it can be hypothesized that intrinsic resistance of NSCLC could be derived from the cell detoxification mechanisms present in normal respiratory epithelial cells, comparative studies between normal respiratory cells and NSCLC cells would be worthwhile. Moreover, although LRP has been detected in lung cancer (Izquierdo et al. 1996a), its role in NSCLC is still a subject of controversy (Berger et al. 2000; Harada et al. 2003).
The present study was therefore designed first to precisely describe and compare the intracellular localization of LRP expression in normal bronchial cells and lung cancer cells from NSCLC samples and second to quantitatively compare the level of expression of LRP in these cells. Moreover, as a recent study from our laboratory showed that DNA aneuploid carcinomatous cells from NSCLC with an increased chromosome 16 number, harboring the mrp1 locus (16p13.1) close to the lrp locus (16p11.2), exhibited higher MRP1 expression than DNA diploid cells (Doubre et al. 2005), we therefore hypothesized that MRP1 and LRP could have a coordinate expression. We also investigated the potential role of the number of lrp gene copies on the level of LRP expression in NSCLC cancer cells.
Materials and Methods
Bronchial Epithelial Cells
Normal bronchial epithelial cells were recovered by bronchial brushings performed for diagnosis in patients suspected of having lung cancer. Bronchial samples (n = 29) containing only cytologically normal epithelial cells were frozen in DMSO and stored at −80C until flow cytometry (seven samples), immunocytochemistry (four samples), confocal microscopy (11 samples), and FISH (seven samples). Samples were collected according to French legislation and the ethical rules of our institution at the time of experimentation.
Tumor Samples
NSCLC specimens, collected according to French legislation and the ethical rules of our institution at time of the experiment, were obtained from 14 patients after surgery performed in the Department of Thoracic Surgery (Dr. Bazelly; Hôpital Tenon, Paris, France). Tumor specimens were snap frozen in liquid nitrogen and stored at −80C. No patient had received chemotherapy before surgery.
Before tissue dissociation for flow cytometry, at least four sets of touch prints were systematically prepared, one stained by the May Grünwald Giemsa method for identification of carcinomatous cells, two others air dried overnight at room temperature and stored at −20C for immunocytochemistry and confocal microscopy, and one fixed with 3:1 methanol/acetic acid for 30 min and stored at −20C for FISH analysis. For flow cytometry, cells were mechanically dissociated from tissues by scraping of defrosted samples in 5% PBS–fetal calf serum (FCS). After PBS washes, aliquots of 4 × 106 cells were stained for flow cytometry analysis.
A549 Cell Line
The NSCLC A549 cell line (American Type Culture Collection; Rockville, MD), known to spontaneously strongly express the LRP protein, was used for controls (Trussardi et al. 1998). A549 cells were expanded in DMEM (Invitrogen; Cergy Pontoise, France) supplemented with 10% (v/v) FCS (Invitrogen), 5 mM glutamine (Sigma; Saint Quentin Falavier, France), 50 U/ml penicillin–streptomycin (Sigma), and 20 μM Hepes (Invitrogen) at 37C in a humidified atmosphere containing 5% CO2.
Antibodies and Reagents
Intracytoplasmic LRP was detected after incubation with monoclonal antibody (MAb) LRP-56 (Tebu-Bio; Le Perray en Yvelines, France) recognizing an intracellular epitope (Scheper et al. 1993) at different final working concentrations according to the method used. Incubation with a mouse IgG2b (Immunotech; Marseille, France) at the same protein concentration as MAb LRP-56 was used as the isotypic control. Secondary antibodies were a biotinylated rabbit anti-mouse F(ab′)2 (Dako; Trappes, France) for immunocytochemistry and a FITC-conjugated F(ab′)2 goat anti-mouse IgG (H&L; Immunotech) for confocal microscopy and flow cytometry.
To check the MAb LRP-56 specificity, immunoprecipitation was performed in five distinct experiments on cell lysates from A549 and two mesothelioma cell lines (BLA and CORO; kindly provided by Dr. M.C. Jaurand, INSERM U674, Paris, France). Approximately 107 cells were processed as previously described (Meschini et al. 2002). Supernatants supplemented with 1% non-fat milk powder, precleared with 100 μl Protein G Sepharose (diluted 1:1 in PBS; Amersham Bioscience, Orsay, France) were spun at 10,000 × g for 10 min at 4C and incubated with 4 μg of undiluted MAb LRP-56 (100 μg/ml) or an equivalent amount of an irrelevant mouse IgG2b immunoglobulin for 2 hr at 4C. Immune complexes were precipitated by incubation with Protein G Sepharose (Amersham Bioscience), washed twice with lysis buffer containing 2% BSA and four times with PBS, taken up in 50 μl of sample buffer (200 mM Tris–HCl, pH 6.8, 1% 2-mercaptoethanol, 8% SDS, 10% glycerol, and 0.05% bromophenol blue), and microfuged. Supernatants were analyzed on 10% PAGE (125 V, 600 mA, 2 hr 30 min) after staining with Coomassie Brilliant Blue G (Sigma). After drying, gels were scanned with a model GS 800 (Bio-Rad; Marnes la Coquette, France). Molecular mass standards were obtained from Invitrogen.
Immunocytochemistry and Confocal Microscopy Analysis
Cytospin preparations of normal bronchial cells and A549 cells were prepared for immunocytochemistry, whereas touch prints were used for tumor cells from NSCLC samples, as previously described (Doubre et al. 2005). Slides were air dried overnight at room temperature, rehydrated with PBS for 5 min, and fixed by paraformaldehyde (PFA, 0.5% in PBS, pH 7.4, 25 min at room temperature). After two 10-min washes in PBS, cells were permeabilized with Triton X-100 0.3% in PBS, 30 sec at room temperature (Sigma), washed twice in PBS for 10 min, and incubated with the PBS/30% human AB serum saturation buffer (Abcys; Paris, France) for 30 min at room temperature before incubation with MAb LRP-56 or IgG2b.
For immunocytochemistry and light microscopy, cell preparations were incubated for 1 hr at room temperature with MAb LRP-56 (1/20; 5 μg/ml) or an equivalent amount of mouse IgG2b diluted in PBS/10% human AB serum and subsequently with biotinylated rabbit anti-mouse F(ab′)2 fragments (8.4 μg/ml) for 30 min at room temperature, followed by three 15-min washes in PBS. The conjugate was revealed using peroxidase-conjugated streptavidin (1/500, 30 min at room temperature) followed by three washes in PBS for 15 min (all reagents were obtained from Dako). Peroxidase activity was revealed with DAB (Dako), nuclei were counterstained with Harris hematoxylin (Bayer; Leverkusen, Germany), and the preparations were mounted with Eukitt (Kindler GmbH; Freiburg, Germany) and observed with a microscope (Zeiss Axioplan, Carl Zeiss, Jena, Germany) equipped with a color video camera (JVC KyF50).
For confocal microscopy, cell preparations were incubated for 1 hr with either MAb LRP-56 (1/5; 20 μg/ml) or the mouse IgG2b isotypic control (20 μg/ml). The slides were then incubated with a FITC-conjugated F(ab′)2 goat anti-mouse IgG (H&L) (0.014 mg/ml) for 30 min in the dark. Incubations with primary and secondary antibodies were performed at room temperature in PBS containing 10% human AB serum. Nuclei were counterstained with DRAQ-5 (1/200 in PBS; Biostatus, Leicestershire, UK) for 3 min at 37C. Centrifuge preparations were mounted with fluorescent mounting medium (Dako) and examined under a confocal laser-scanning microscope (Leica; Wetzlar, Germany). Cells were viewed in the X-Y or Y-Z plane, and the images were recorded.
For colocalization of LRP and actin, cells grown on Lab-Tek chamber slide system (ATGC; Marne la Vallee, France) with an ∼90% confluence were used. Cells were fixed for 15 min by PFA (1% in PBS, pH 7.4, 25 min at room temperature). After a PBS wash, cells were permeabilized with 0.3% Triton X-100 in PBS for 30 sec and washed three times for 10 min each with PBS. Cells were then incubated with tetramethylrhodamine B isothiocyanate-labeled phalloidin (1/200; Sigma) for 40 min. After three 10-min washes in PBS, cells were then incubated with either MAb LRP-56 (20 μg/ml) or the mouse IgG2b isotypic control (20 μg/ml). Further LRP staining procedures were performed as described above.
Flow Cytometry Analyses
Cells were double stained with MAb LRP-56 and propidium iodide (PI) to concomitantly evaluate the intracellular expression of LRP and DNA ploidy status. Cells were washed three times with PBS, and the pellet (5 × 105 cells in 100 μl PBS) was fixed in 0.5% PFA in PBS for 10 min at room temperature. After centrifugation for 5 min at 1400 × g at 4C and blotting of residual PFA, cells were permeabilized by adding saponin (Intraprep kit 100 μl; Immunotech) for 5 min at room temperature. Cells were then incubated overnight at 4C either with MAb LRP-56 (4 μg/ml) or the isotypic control, a mouse IgG2b (4 μg/ml). After PBS washes and centrifugation, the cell pellet was incubated with a FITC-conjugated F(ab′)2 goat anti-mouse IgG (H&L) (0.028 mg/ml) for 30 min in the dark at room temperature. After one PBS wash, the cell pellet was suspended in 1 ml PI (50 ng/ml) containing RNase (DNA prep kit; Beckman Coulter, Villepinte, France) and kept in the dark at 37C for 30 min prior to flow cytometry analysis.
For flow cytometry analysis, samples were run on a Coulter EPICS Elite flow cytometer (Beckman Coulter) equipped with a single argon ion laser. Excitation was performed at 488 nm, and the emission filters used were 635 nm (red, PI) and 525 nm (green, FITC). At least 2 × 104 cells per sample were analyzed, and data were stored in list mode files. LRP-positive cells were determined by reference to the negative level of the isotypic control and gated on dual parameter dot-plot LRP staining (green fluorescence) against side scatter (excluding cells with a high level of autofluorescence, such as macrophages).
Mean fluorescence index (MFI) was defined as the ratio of the mean fluorescence intensity obtained after incubation with MAb LRP-56 and after incubation with the isotypic control. This index was used for semiquantitative evaluation of LRP cell expression in cell types and to compare the levels of LRP expression in DNA diploid vs DNA aneuploid cells, as previously described (Doubre et al. 2005). DNA ploidy and S-phase data were analyzed using Wincycle software (Phoenix; San Diego, CA). DNA ploidy was analyzed according to the guidelines of the 1992 Cytometry Consensus Conference (Shankey et al. 1993). DNA ploidy was classified using a DNA index (DI), i.e., the ratio of the channel peaks between the studied cell population and the diploid cells. A sample was defined as DNA diploid when all the cell population has a DI between 0.95 and 1.05 and DNA aneuploid when at least one cell population with DI ≤0.95 or ≥1.05 was observed in at least 5% of the cells (Doubre et al. 2005).
This analysis was performed on bronchial cells (seven samples) and on tumor cells obtained from NSCLC (14 samples), as well as on A549 cell line (from three different experiments) for control.
FISH
Dual-color FISH analysis on interphasic nuclei was performed with a mixture of spectrum green-labeled lrp gene probe and a rhodamine-labeled chromosome 16 α-satellite centromere probe (Amplitech; Compiegne, France), which allowed simultaneous determination of a number of lrp gene copies and chromosome 16. The bacterial artificial chromosome (BAC clone RP11-279M12 from the RP11 human BAC clone library mapping to 16p11.2 at lrp locus; Wellcome Trust Sanger Institute, Cambridge, UK) was used for lrp gene hybridization. Isolation and purification of DNA were performed using the Qiagen Hispeed Plasmid Maxi Kit (Qiagen; Courtaboeuf, France) according to the manufacturer's instructions. BAC DNA was labeled with spectrum green dUTP by nick translation (Vysis; Maurence Scopont, France). This probe was purified by ethanol precipitation and dissolved in 1/10 TE buffer, pH 8.
Dual-color FISH was performed on cytospin preparations of normal bronchial cells (seven samples) and A549 tumor cell line and on touch imprints from 14 NSCLC samples. Cells were fixed and incubated with the mixture of directly labeled lrp gene probe (spectrum green) and a directly labeled chromosome 16 centromeric probe (rhodamine) [1 μl of chromosome 16 α-satellite centromere probe and 5 μl of BAC DNA (50 μg/ml) in 4 μl of LSI hybridization buffer (Vysis)].
Briefly, slides were treated in a pepsin solution (50 ng/ml, 0.01 N HCl; Sigma) for 15 min at 37C; dehydrated in a chilled alcohol series of 70%, 80%, and 100% for 2 min each; and hybridized with 10 μl of the hybridization mixture applied onto the slide. Denaturation was then performed at 72C for 2 min. Hybridization was performed overnight in a dark moist chamber at 37C. Slides were subsequently washed in 0.4X SSC at 73C for 2 min and in 2X SSC, 1% Tween 20, pH 7, at room temperature for 2 min. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) in Vectashield (Abcys).
Slides were examined with a DM400B Leica epifluorescence microscope equipped with filter sets optimized for DAPI, spectrum green, and rhodamine. Images were captured with a Zeiss microscope and a cooled CCD camera using Genikon Imaging System (Alphelys; Plaisir, France). Same-colored signals were counted as two if they were separated by at least one signal diameter and had the same intensity, size, and shape. Hybridization signals were scored in at least 100 intact non-overlapping nuclei.
Statistical Analysis
Results are expressed as mean ± SD. Statistical analysis was performed with Statview Software (Abacus Concept; Berkeley, CA). Non-parametric tests were applied for the various data comparisons: a paired test (Wilcoxon) to compare values from individual specimens and a non-paired test (Mann-Whitney). Statistical significance was estimated when p value ≤0.05.
Results
Immunoprecipitation With MAb LRP-56
Immunoprecipitation results of cell lysates from cell lines with MAb LRP-56 are shown in Figure 1. A band at the expected size of LRP, i.e., 110 kDa (Scheper et al. 1993; Izquierdo et al. 1996a) was clearly demonstrated after precipitation with MAb LRP-56 and was absent after precipitation with the mouse IgG2b immunoglobulin. These results confirmed the specificity of MAb LRP-56 to detect human LRP, as already reported (Scheper et al. 1993; Izquierdo et al. 1996a).
Immunocytochemistry and Light Microscopy
LRP was immunodetected, after incubation with MAb LRP-56, in normal bronchial epithelial cells and in tumor cells from NSCLC samples as well as in the A549 control cell line. A similar granular intracytoplasmic pattern of LRP localization was observed in these various cell types. The percentages of positive cells immunolabeled were within the same ranges in normal bronchial cells (84 ± 13.2%) and tumor cells from NSCLC samples (93 ± 2.9%) as in the A549 cell line (88 ± 4.8%). No staining was observed after incubation with the mouse IgG2b isotypic control in any of the cell types (Figure 2).
Immunofluorescence and Confocal Microscopy
Immunolabeling was observed in most cells regardless of cell type. Confocal microscopy confirmed the intra-cytoplasmic localization of LRP (Figure 3). In bronchial cells (Figure 3A), granular staining was observed in the apical and basal parts of the cells. In cells from NSCLC samples (Figure 3B), a similar granular staining was observed throughout the cytoplasm as in the A549 cell line (Figure 3C). No perinuclear reinforcement or NPC staining was observed. Double-staining colocalizing LRP and actin confirmed that the cytoplasmic distribution of LRP was not driven by the actin microfilament network (Figure 3C).
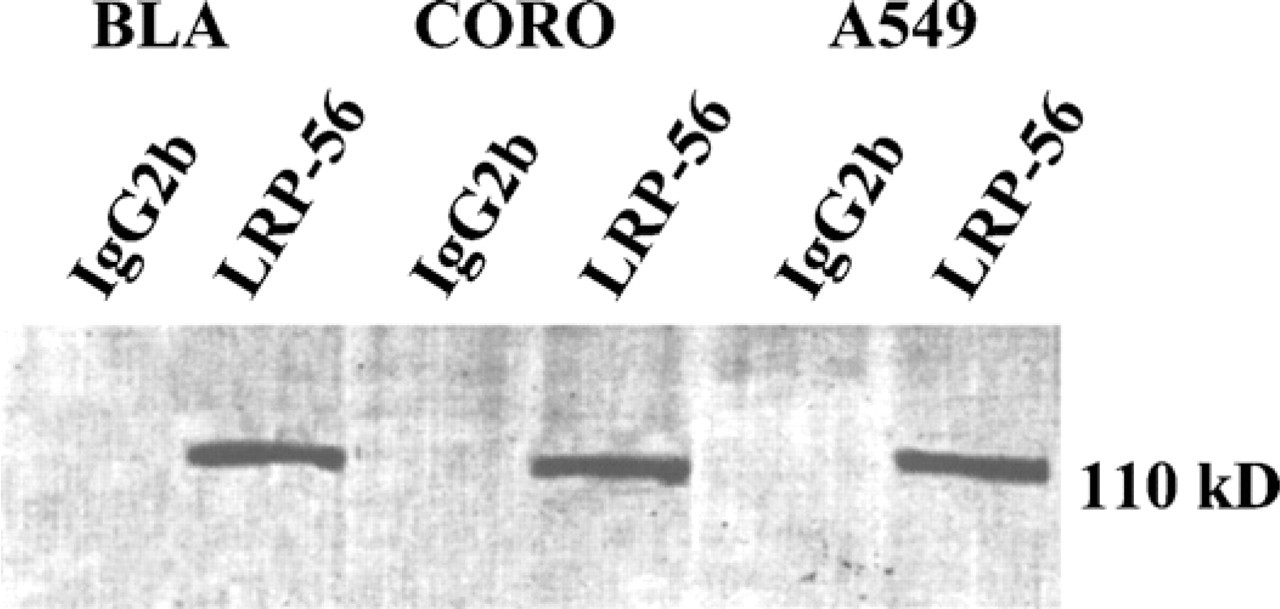
Immunoprecipitation with monoclonal antibody (MAb) LRP-56. Immunoprecipitation of BLA, CORO, and A549 cell lysates using MAb lung resistance-related protein (LRP)-56. Lysates were immunoprecipitated as described in Materials and Methods with LRP-56 or a mouse IgG2b. A 110-kDa band is observed in the cell lysates from the three cell lines. No band was detected after precipitation with the mouse IgG2b.
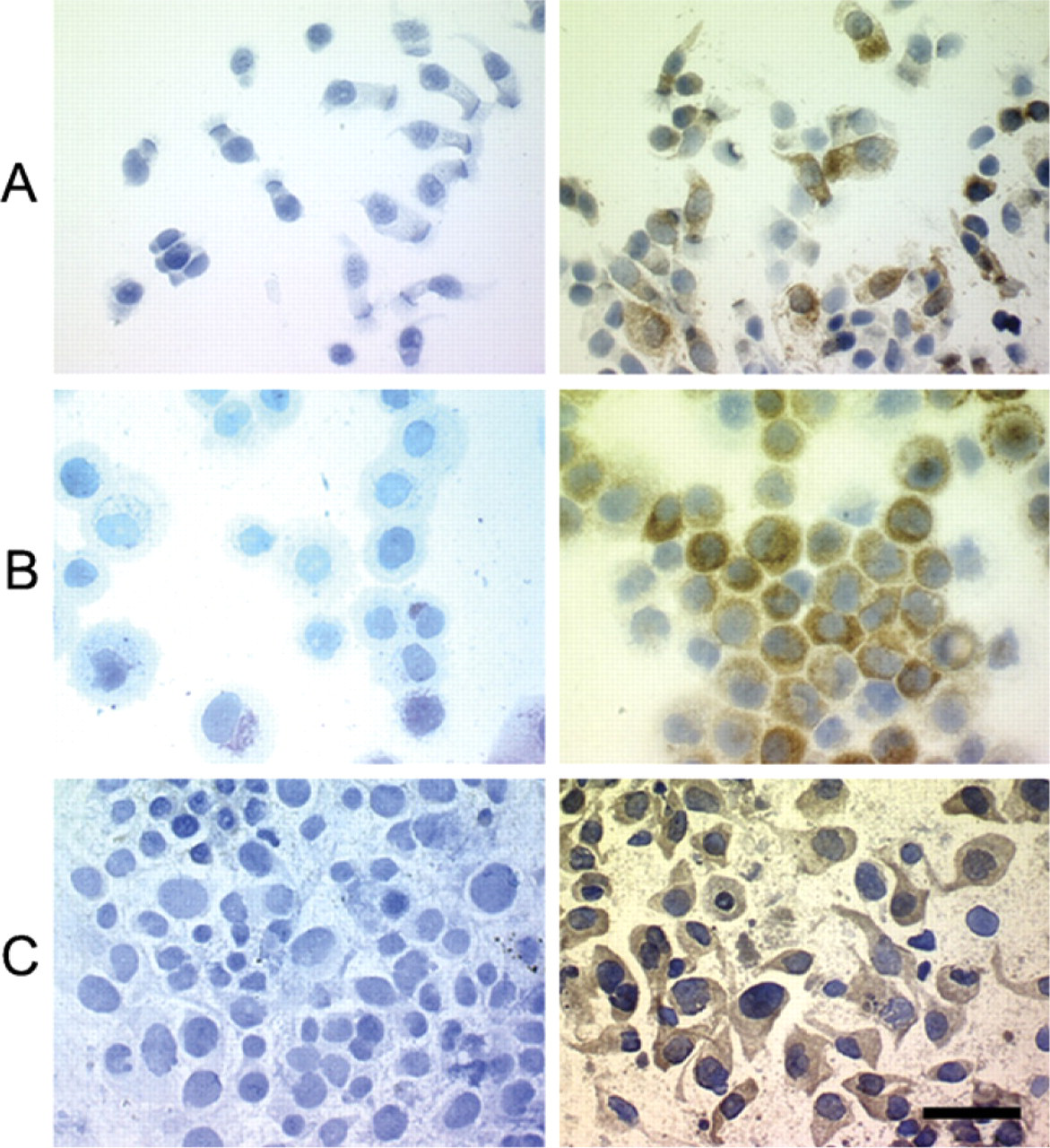
Immunodetection of LRP by light microscopy. Immunostaining of LRP with MAb LRP-56 (5 μg/ml) using immunoperoxidase in normal bronchial epithelial cells (
Flow Cytometry Analyses
MFI used for semiquantitative evaluation of LRP expression was 22 ± 6.7 in normal bronchial cells (seven samples) and 29.37 ± 15.2 in cells obtained from NSCLC samples (14 samples), similar to A549 cell line (20 ± 11.3). No significant difference in LRP expression was detected between tumor cells from NSCLC and normal bronchial cells (p = 0.29) (Figure 4).
After DNA labeling, cells from the seven bronchial samples were all DNA diploid, in contrast to A549 cells that were all DNA aneuploid. Of the 14 samples from NSCLC, three consisted of DNA diploid cells, whereas 11 contained at least one DNA aneuploid cell subpopulation (Table 1).
MFI of tumor cells examined as a whole within the 14 samples of NSCLC was 29.37 ± 15.2. However, this index was not significantly different when comparing the 11 samples with DNA aneuploid cells (33.5 ± 14.9) to the three samples containing only DNA diploid cells (25.2 ± 15.5) (p = 0.49). Moreover, when separately analyzing the diploid and aneuploid cell subpopulations that coexisted within DNA aneuploid samples, an index of 31.8 ± 13.3 was observed in DNA aneuploid cells, not significantly different from that observed in DNA diploid cells (25.12 ± 17.06, p = 0.11) (Figure 4). Analysis of LRP expression after LRP-PI double staining is summarized in Table 1.
FISH Analysis
FISH analysis of chromosome 16 and lrp gene loci performed on normal bronchial cells, A549 cell line, and tumor imprints was correlated with DNA ploidy (Figure 5; Table 1). After dual-color FISH of interphasic nuclei with chromosome 16 α-satellite centromere probe and BAC DNA clone RP11-279M12, the number of copies of lrp gene loci and chromosome 16 centromere per interphasic nucleus was always similar, regardless of the cell type studied.
All bronchial cells were DNA diploid, and FISH studies revealed that 96–99% (mean 98 ± 0.8%) of these cells were disomic for chromosome 16. In contrast, in the DNA aneuploid A549 cells, 87–96% (mean 91.3 ± 4.5%) were trisomic for chromosome 16 (Table 1).
Among the 14 samples from NSCLC, 3 tumors were DNA diploid and 11 tumors DNA aneuploid. In DNA diploid tumors, an average of 75.7 ± 38.2% of tumor cells exhibited two signals for chromosome 16. In contrast, intracellular variations in the chromosome 16 copy number were observed in DNA aneuploid tumors with an average of 68 ± 24.1% of tumor cells with more than two copies of chromosome 16 (Figure 5 and Figure 6; Table 1). The most frequent anomalies were three or more than three copies of chromosome 16.
Discussion
It can be hypothesized that an intrinsic chemoresistance of NSCLC could be derived from the cell detoxification mechanisms present in normal respiratory epithelial cells and LRP to be at least partly responsible for the MDR mechanisms of cell detoxification and tumor cell resistance to chemotherapy. Therefore, the present study was designed first to precisely describe and compare its intracellular localization in normal and cancer respiratory cells from NSCLC and second for the first time to quantitatively compare its level of expression in these different cells and in relation to chromosomal disorders. A first step in this study, using immunocytochemistry, clearly demonstrated that LRP is expressed with a similar pattern in normal bronchial cells and in NSCLC cells. As shown by confocal microscopy, LRP had a diffuse intracytoplasmic pattern of localization in normal bronchial epithelial cells as in cancer cells obtained by tissue imprints from NSCLC samples. This observation is in accordance with previous studies on other cell types reporting that most vaults have a cytoplasmic localization (Kedersha and Rome 1986; Scheper et al. 1993; Berger et al. 2000). LRP has been described to be associated not only with cytoplasmic organelles, but a small percentage of vault proteins (∼5%) are also localized in the nuclear membrane and nuclear pore complex (NPC), supporting the hypothesis that vaults could act as shuttles between the nucleus and cytoplasm via NPC (Hamill and Suprenant 1997; Slesina et al. 2005). However, nuclear localization of MVP has been shown using cell fractionation and/or electron microscopy methods (Chugani et al. 1993; Slesina et al. 2005), particularly sensitive to show a small amount of protein. The role of MVP as it shuttles between nucleus and cytoplasm is still hypothetical. In vitro studies comparing the cellular localization of MVP before and after drug exposure did not show movement from cytoplasm to nucleus (Berger et al. 2000; van Zon et al. 2006), but an accumulation at the nuclear envelope has been observed by van Zon et al. (2006). However, these results cannot exclude a role of MVP in signal transduction. We did not observe this type of NPC labeling in any cell type, and our study concerned normal epithelial cells and tumor cells not exposed to chemotherapy. Our results are, therefore, in favor of an exclusive intracytoplasmic role of vaults and LRP in normal cells and NSCLC tumor cells. Moreover, LRP/vaults have been shown to colocalize with filamentous actin in the neuritic tips of neuron-like P12 cells (Herrmann et al. 1999). This type of colocalization, demonstrated to be involved in intracellular vault movement, was not detected in our experiments in NSCLC cells after double staining with actin, in line with the results reported by Berger et al. (2000).
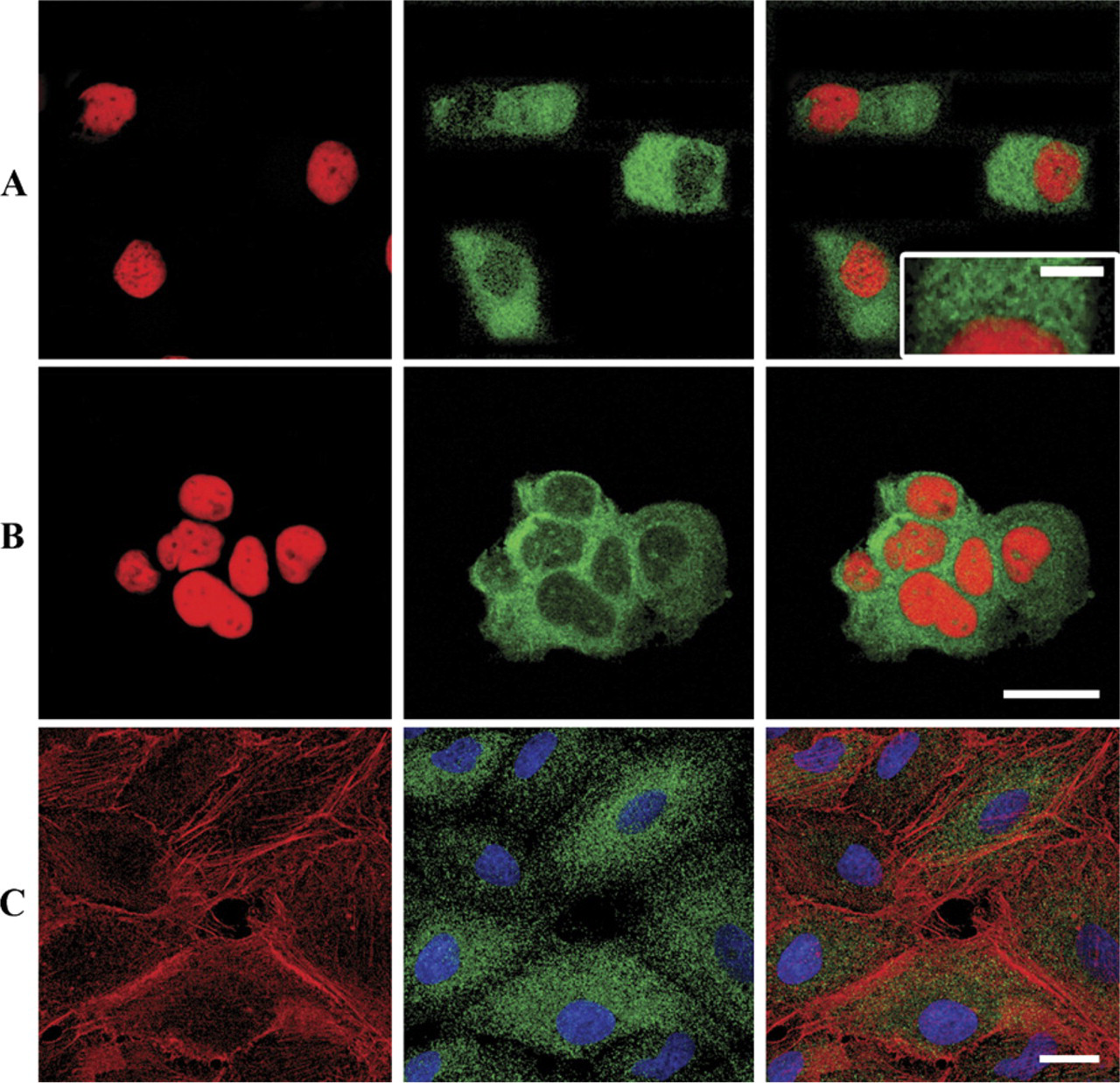
Confocal microscopy of LRP in normal bronchial epithelial cells, non-small cell lung cancer (NSCLC), and A549 cells. Illustrations of indirect immunofluorescence analyzed on horizontal (x-y axis) optical sections after incubation with MAb LRP-56. (
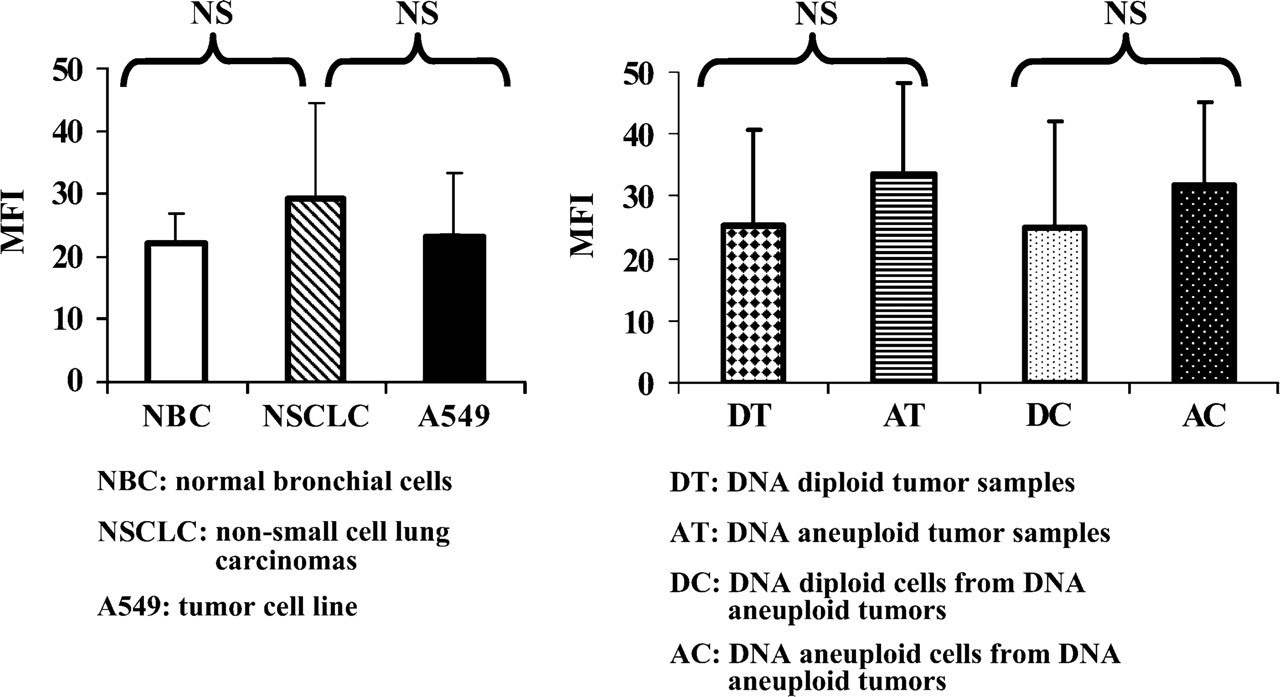
Mean fluorescence index (MFI) of LRP in various cells analyzed by flow cytometry. (Left panel) MFI of LRP (mean ± SD) evaluated on seven normal bronchial cells samples, 14 tumor cells from NSCLC, and the A549 tumor cell line. No significant differences (NS) in the levels of LRP protein expression were detected between normal bronchial cells, the A549 tumor cell line, and tumor cells from NSCLC. (Right panel) LRP MFI (mean ± SD) evaluated on cells obtained from three DNA diploid and 11 DNA aneuploid tumor samples. Columns on the right show results restricted to the DNA diploid and DNA aneuploid cell subpopulations from DNA aneuploid tumors. No significant difference (NS) in LRP protein expression was detected between DNA diploid tumors and DNA aneuploid tumors or between DNA diploid and DNA aneuploid cell subpopulations from DNA aneuploid tumors.
Results of DNA ploidy, LRP MFI, and chromosome 16 and lrp gene copy numbers
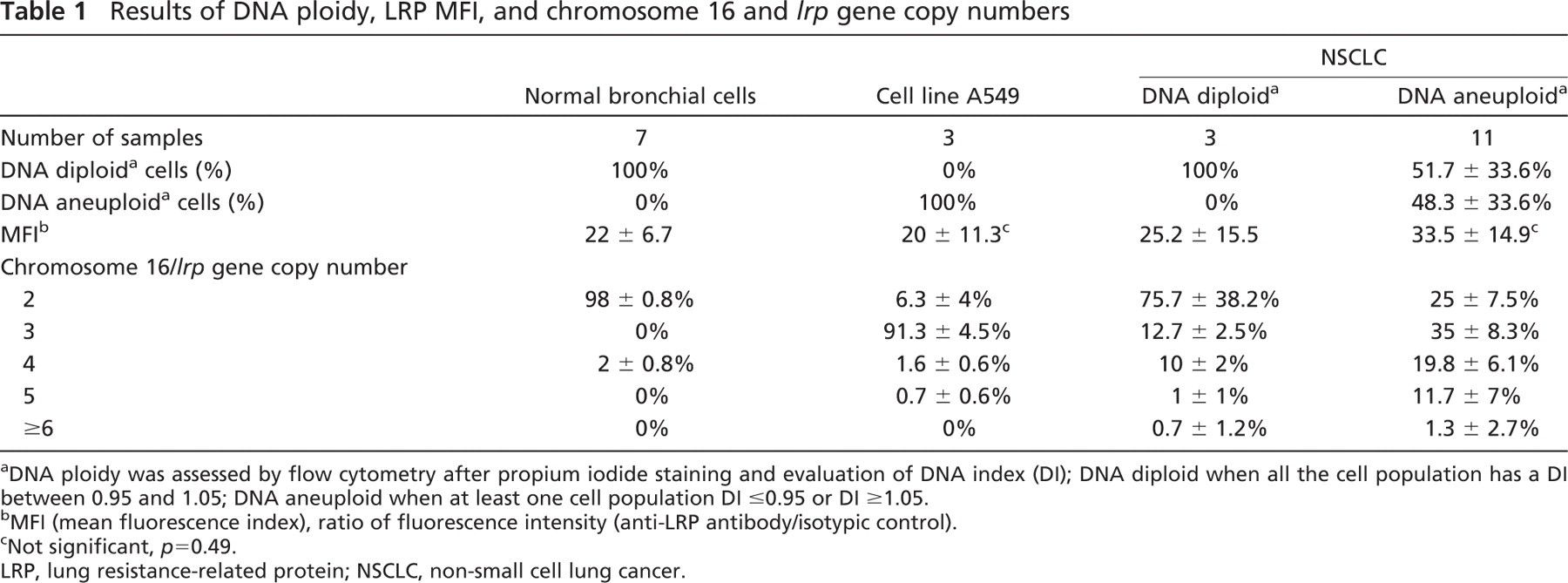
DNA ploidy was assessed by flow cytometry after propium iodide staining and evaluation of DNA index (DI); DNA diploid when all the cell population has a DI between 0.95 and 1.05; DNA aneuploid when at least one cell population DI ≤0.95 or DI ≥1.05.
MFI (mean fluorescence index), ratio of fluorescence intensity (anti-LRP antibody/isotypic control).
Not significant, p = 0.49.
LRP, lung resistance-related protein; NSCLC, non-small cell lung cancer.
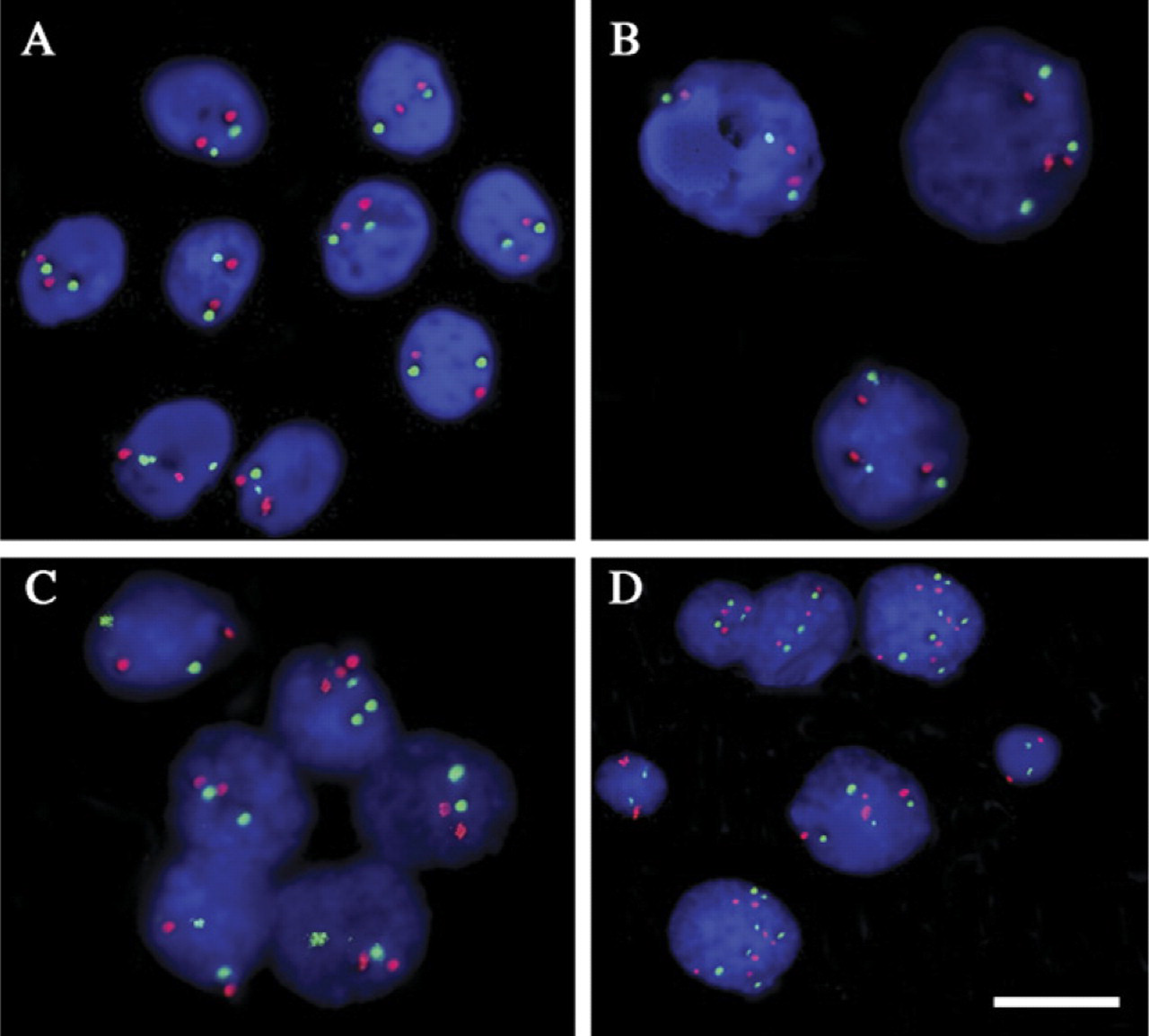
Examples of FISH results after cohybridization of interphasic nuclei with a chromosome 16 centromeric probe (red signals) and BAC DNA clone RP11-279M12 (lrp gene on 16p11.2, green signals). (
The second step of this study was to use a flow cytometry method for semiquantitative evaluation of LRP expression. Most of the studies concerning LRP expression in solid tumors were based on immunohistochemistry using a score evaluation (Dingemans et al. 1996; Volm et al. 1997) or reverse transcription–polymerase chain reaction (RT-PCR) evaluating LRP mRNA (Ohno et al. 2001), whereas the majority of studies on hematological malignancies used flow cytometry for LRP detection (Damiani et al. 2004). Moreover, this method has not been previously used to compare LRP expression in normal and carcinomatous cells from the same tissues. We previously reported the use of such a method for evaluation of MRP1 expression in normal bronchial and NSCLC cells (Doubre et al. 2005). LRP expression was found in 100% of NSCLC cells, in line with the results reported by Berger et al. (2000) who evaluated LRP expression by mRNA detection. However, the rate of LRP-positive cells evaluated by flow cytometry is higher than that previously reported for LRP expression evaluated by immunohistochemistry (Dingemans et al. 1996; Volm et al. 1997; Harada et al. 2003; Raybarova et al. 2004). This difference can probably be explained by a higher sensitivity of flow cytometry analysis compared with immunohistochemistry (Leers et al. 2002). Similarly, LRP was detected in bronchial epithelial cells in our study as in other studies (Nooter et al. 1995), suggesting a role of LRP in the cell detoxification process for cells localized at the air–tissue interface (Dingemans et al. 1996; Izquierdo et al. 1996a) where detoxification mechanisms are particularly well developed. A similar level of LRP expression was observed in bronchial cells and carcinomatous cells from NSCLC samples, similar to the A549 cell line known to spontaneously have a high level of LRP expression (Trussardi et al. 1998). This is at variance with observations in the colon, where LRP is expressed in the surface epithelium and upper part of the crypts of normal colorectal epithelium (Dingemans et al. 1996; Meijer et al. 1999), but a definitive and abrupt increase of LRP levels is observed early in the process of colorectal carcinogenesis, from normal epithelium to adenoma and from adenoma to carcinoma (Meijer et al. 1999). Some types of carcinomatous cells have been reported to express significantly higher levels of LRP than their normal counterparts (e.g., thyroid carcinoma), whereas LRP expression appears to be downregulated during malignant transformation in other tumors (e.g., germ-cell tumors) (Izquierdo et al. 1996a). These results suggest that a similar level of LRP expression was maintained in epithelial respiratory cells throughout the carcinogenesis process.
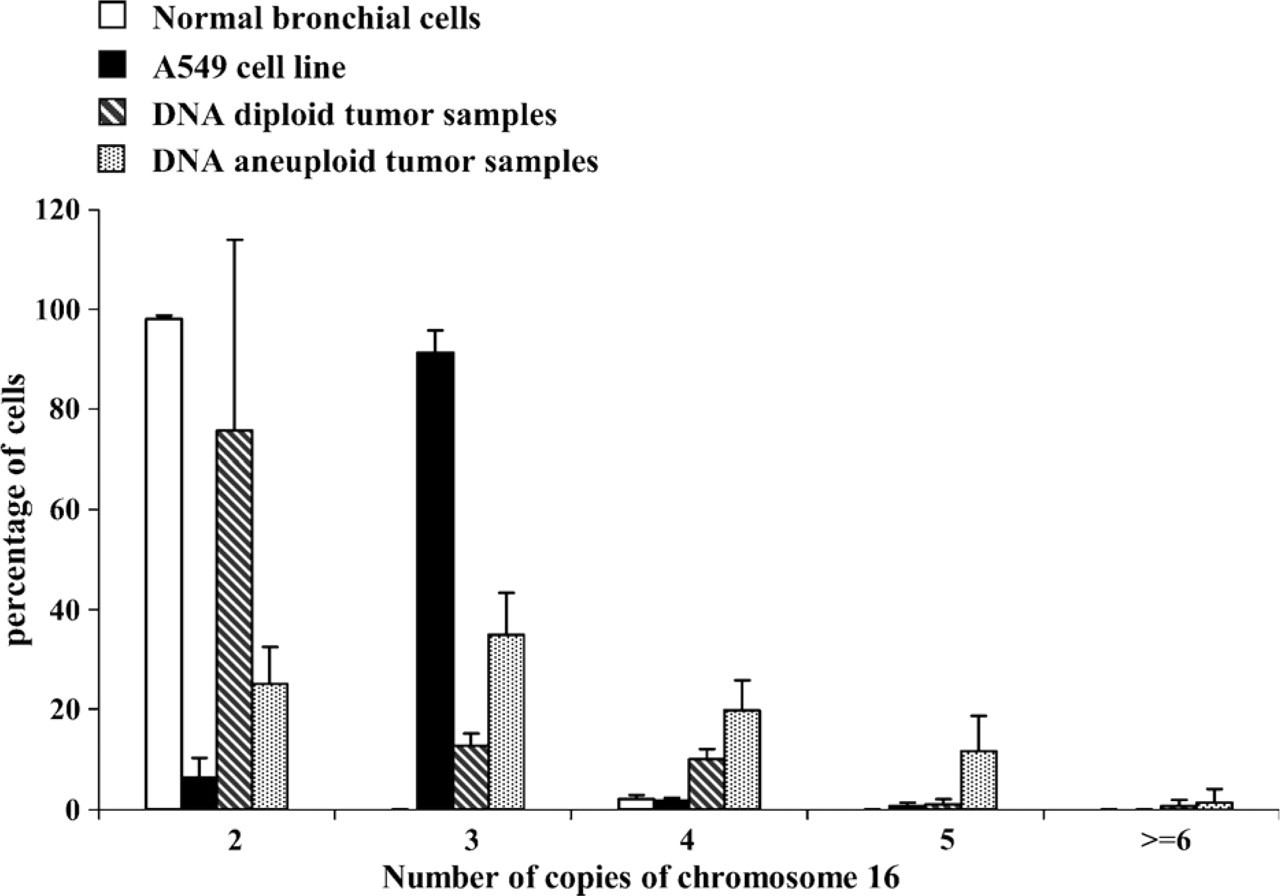
Distribution of the chromosome 16 and lrp gene copy numbers per 100 cells (mean ± SD) observed after FISH analysis in relation to DNA ploidy status in seven samples of normal bronchial cells, three samples of A549 cells, and in three DNA diploid and 11 DNA aneuploid tumor samples.
The hypothesized role of LRP in resistance to chemotherapy is still a subject of controversy. However, the role of LRP as a prognosis factor in cancer is probably not restricted to the MDR phenotype, MVP being associated with several cellular processes also involved in cancer development like cell proliferation, motility, differentiation, or cell signaling (Steiner et al. 2006). Clinically, LRP overexpression has been associated with poor responses to chemotherapy in different malignancies such as NSCLC (Izquierdo et al. 1995; List et al. 1996; Volm et al. 1997; Raaijmakers et al. 1998). Similarly, LRP has been shown to be overexpressed in a subgroup of cisplatin-resistant NSCLC cell lines (Berger et al. 2000), and LRP has recently been reported to be a predictive marker for treatment response in NSCLC and testicular germ-cell tumors (Harada et al. 2003; Mandoky et al. 2004). However, other studies failed to detect such an association (Dingemans et al. 1996), and another study showed that LRP expression in NSCLC was not enhanced in patients treated with chemotherapy (Berger et al. 2005). However, it has been shown that the other lung cancer subtype, SCLC, which is initially very sensitive to chemotherapy, displays very low LRP expression (Dingemans et al. 1996).
We recently showed that DNA aneuploid NSCLC carcinomatous cells with an increase in chromosome 16 number exhibited a higher MRP1 expression than DNA diploid cells (Doubre et al. 2005). As the lrp gene is localized on the short arm of chromosome 16 (16p11.2), 27 centimorgan or an estimated 18 mega-bases proximal to the mrp1 gene (16p13.1), we tested the hypothesis that LRP and MRP1 could both be concomitantly overexpressed in the case of chromosome 16 polysomy. However, we found a similar level of LRP expression in DNA diploid and DNA aneuploid NSCLC cells as in normal bronchial epithelial cells, regardless of the chromosome 16 number. This dissociation between LRP and MRP1 expression is at variance with the results described in ovarian carcinoma (Yakirevich et al. 2006). However, several studies in leukemia and unselected human cancer cell lines (Izquierdo et al. 1996b; Michieli et al. 1997) have provided evidence that MRP and LRP expression are not coregulated. In our study, the increase of chromosome 16 copies was associated with DNA aneuploidy but not with an increase in the level of LRP protein expression. Similarly, differences in the expression of LRP mRNA and protein have been described in other studies (Laurencot et al. 1997), suggesting a strong posttranscriptional regulation of LRP expression. Our results do not support the gene dosage hypothesis for LRP expression control. Moreover, no lrp overexpression was observed in NSCLC cells, in line with the results reported by Berger et al. (2005). These results suggest that the LRP expression observed in NSCLC is controlled by mechanisms different from those involved in MRP1 expression, whereas both proteins are associated with the MDR phenotype.
In conclusion, in the present study LRP expression, which is associated with a ubiquitous role of vaults in cell detoxification processes, was found in NSCLC carcinomatous cells from tumor samples similar to that observed in normal bronchial epithelial cells. No alteration of the LRP intracellular localization and/or level of expression was observed when considering the normal and carcinomatous subsets of cells from the same tissue, i.e., respiratory airways. Moreover, on the basis of these results, we can therefore hypothesize that the level of LRP expression observed in NSCLC is maintained through the carcinogenesis process of respiratory cells and not altered by the increased number of copies of chromosome 16 catalyzed by cell aneuploidy.
Footnotes
Acknowledgements
This study was supported by “Legs POIX” 2005 and Amis des Centres des Tumeurs de Tenon.
We thank the Department of Thoracic Surgery, particularly Dr. B. Bazelly, and the Department of Pathology (Dr. M. Antoine) for the generous gifts of the NSCLC specimens, and the Department of Respiratory Medicine (Prof. C. Mayaud) for normal bronchial epithelial cells. We are grateful to Prof. J. P. Siffroi, A. Leneveu, S. Giacuzzo, and S. Lucchini for their technical assistance in cytogenetics.