
Abstract
Multiple correlative immunolabeling permits colocalization of molecular species for sequential observation of the same sample in light microscoopy (LM) and electron microscopy (EM). This technique allows rapid evaluation of labeling via LM, prior to subsequent time-consuming preparation and observation with transmission electric miscroscopy (TEM). The procedure also yields two different complementary data sets. In LM, different fluorophores are distinguished by their respective excitation and emission wavelengths. In EM, colloidal metal nanoparticles of different elemental composition can be differentiated and mapped by energy-filtering transmission electron microscopy with electron spectroscopic imaging. For the highest level of spatial resolution in TEM, colloidal metal particles were conjugated directly to primary antibodies. For LM, fluorophores were conjugated to secondary antibodies, which did not affect the spatial resolution attainable by fluorescence microscopy but placed the fluorophore at a sufficient distance from the metal particle to limit quenching of the fluorescence signal. It also effectively kept the fluorophore at a sufficient distance from the colloidal metal particles, which resulted in limiting quenching of the fluorescent signal. Two well-defined model systems consisting of myosin and α-actinin bands of skeletal muscle tissue and also actin and α-actinin of human platelets in ultrathin Epon sections were labeled using both fluorophores (Cy2 and Cy3) as markers for LM and equally sized colloidal gold (cAu) and colloidal palladium (cPd) particles as reporters for TEM. Each sample was labeled by a mixture of conjugates or labels and observed by LM, then further processed for TEM.
Keywords
C
In general, immunolabeling uses unconjugated primary antibodies labeled with secondary or tertiary antibodies conjugated to fluorophores for LM and to electron-dense metal markers detected in EM (Faulk and Taylor 1971). For correlative LM/EM labeling, conjugation of an antibody or active fragment of an antibody to both fluorophores and electron-dense markers would be advantageous. However, direct conjugation of both colloidal metal particle and fluorescent dye to the same antibody molecule results in nearly total quenching of the fluorescent signal (Kandela et al. 2003). This is apparently due to resonance energy transfer (Powell et al. 1997). This is the case for colloidal particles in the small (5 nm) size range as well as for larger particles. However, if the metal particle is conjugated to the primary antibody and the fluorescent dye to a second (anti-primary) antibody, the fluorescent dye is spaced a sufficient distance from the metal particle to substantially reduce quenching. With the fluorophore conjugated to the secondary antibody and colloidal particles to the primary antibody, we found quenching to be reduced by 50% in the case of 5-nm particles and by 20% in the case of 18-nm particles (Kandela et al. 2003; Kandela and Albrecht in press). This level of fluorescent signal is sufficient for most applications, particularly where a CCD camera of reasonable sensitivity is used. Placing the colloidal particles on the primary antibody or primary active antibody fragment, Fab, or on a ligand (or active ligand fragment in the case of ligand labeling) provides the high level of spatial localization required for molecular or submolecular imaging using EM. In this case the colloid particle is localized as closely as possible to the epitope it is being used to identify. The presence of the fluorescent dye on the secondary antibody places it further from the epitope being labeled but still well within the resolution required for most LM applications.
Multiple labeling, the simultaneous labeling and identification of several antigenic species, is greatly facilitated in LM by a variety of dyes covering a wide range of excitation and emission wavelengths. For EM, a common approach to multiple labeling involves the use of colloidal particles of different sizes. This can be problematic for purposes of high-resolution imaging, semi-quantitative measurement of epitope numbers, or where epitope density is high. In these cases, particles in the 5-nm range are preferable for labeling each of the different epitopes.
To address this issue, we have developed particles of similar sizes but having different shapes or different elemental compositions (Meyer and Albrecht 2001, 2002; Kandela et al. 2004, 2005). Shape can be recognized using high-resolution imaging, and particles of different composition can be differentiated by energy-filtering TEM (EFTEM) (Meyer and Albrecht 1999; Bleher et al. 2004). In the current study we have investigated a combination of the latter approach for multiple correlative imaging. We have utilized 6-nm gold (cAu6) or 6-nm palladium (cPd6) conjugated to primary antibodies for epitope identification and localization in EM. Secondary antibody conjugated to different fluorophores, either Cy2 or Cy3, permits identification and localization of epitopes in LM prior to examination of the same specimen in EM.
In this way, maximal spatial resolution in EM is achieved using the primary conjugates, whereas the larger distance between secondary fluorophore conjugates and the actual antigenic binding site does not influence the generally lower spatial resolution of LM. Different fluorophores can be distinguished by choosing appropriate filter combinations with dye-specific excitation and emission wavelengths, whereas cAu6 and cPd6 can be mapped with electron spectroscopic imaging (ESI) in an EFTEM.
Materials and Methods
Preparation of Colloids
cAu6 was produced by reducing HAuCl4 with white phosphorus (Faulk and Taylor 1971). To 240 ml double-distilled water (ddH2O), 0.75 ml of 4% HAuCl4 was added. The pH was adjusted to 7.0–7.2 using K2CO3 (0.2 N). For the reduction reaction, 2 ml of saturated phosphorous solution, prediluted 1:5 in ether, was used. The solution was swirled gently in a boiling flask for 15 min and boiled with reflux for 20 min.
For preparation of cPd6, K2PdCl4 was reduced with sodium citrate and sodium ascorbate (Meyer and Albrecht 2003). Two hundred μl of freshly prepared 40% sodium citrate and 1.64 ml of 1% K2PdCl4 were added to 93 ml double-distilled water, and the pH was adjusted to 7.4 with K2CO3 (0.2 N). The solution was heated until boiling, and 5 ml of 4% hot sodium ascorbate was added. Boiling with reflux was continued for 30 min.
Conjugation
cAu6 was conjugated to monoclonal mouse anti-myosin (Sigma-Aldrich; St Louis, MO) or to rabbit anti-α-actinin antibodies (Novus Biologicals; Littleton, CO) at pH 7.4, adjusted with 0.2 N K2CO3. The minimum amount of antibody required to stabilize the colloid was determined with a concentration gradient (Horisberger and Rosset 1977). cAu6 conjugates were collected by ultracentrifugation at 20,000 × g (Ti 50.2 rotor, L5-50 Beckman Ultracentrifuge; Beckman Coulter, Fullerton, CA) for 30 min. Soft pellets were resuspended in 0.05 M HEPES buffer, pH 7.4. Conjugation of cPd6 to mouse anti-actin (Sigma) or rabbit anti-α-actinin antibodies was performed at pH 8.5–9. After the minimum amount of antibodies required to stabilize the cPd6 was determined, cPd6 conjugates were sedimented at 15,000 × g for 30 min and resuspended in 0.05 M HEPES buffer (pH 7.4).
Sample Preparation
Skeletal Muscle Tissue. Rat skeletal muscle tissue was utilized as a model for labeling based on the well-defined location of proteins within sarcomere. Small pieces of rat skeletal muscle tissue were washed briefly in PBS, pH 7.4, and fixed for 2 hr in 4% freshly prepared formaldehyde and 0.1% glutaraldehyde in 0.1 M phosphate buffer (PB), pH 7.4. Samples were rinsed in PBS and in 0.05 M glycine in PBS for 20 min each. Specimens were then dehydrated in 30%, 50%, 70%, 90%, and 2 × 100% ethanol for 20 min each. After two washes with propylene oxide, tissue pieces were infiltrated with propylene oxide. Epon mixtures of 2:1 and 1:1 for 2 hr each were used with a 1:2 mixture overnight (Embed 812; EMS, Hatfield, PA). Specimens were transferred to fresh resin for 2 × 3 hr and positioned in embedding molds in fresh resin. All steps were performed at room temperature. Epon was polymerized at 65C for 48 hr.
Platelets. As with muscle tissue, platelets provide a well-defined model system relative to distribution of cytoskeletal proteins. Human platelets were purified, spun down at 500 × g and resuspended in HEPES–Tyrodes buffer, pH 7.4 (Olorundare et al. 1992). After spinning again, pellets were fixed in 2% paraformaldehyde, 0.1% glutaraldehyde, and 0.1% tannic acid in 0.1 M phosphate buffer (PB) overnight at 4C. Fixed pellets were resuspended in small volumes of 2% melted agarose. Once the agarose was solidified, pellets were cut into small pieces and rinsed in PBS two times for 10 min and in 0.05 M glycine in PBS for 2 hr. Then, samples were dehydrated and embedded as described for skeletal muscle samples. Both rat muscle tissue and human platelets were obtained with the approval of appropriate institutional review boards, Research Animal Resources, and Human Subjects, respectively, at the University of Wisconsin, Madison.
Correlative Immunolabeling
For immunolabeling, ultrathin 60-nm sections were cut with a diamond knife in an ultramicrotome (RMC Cryosystems; Tucson, AZ) and collected on 400 mesh finder grids (Ted Pella; Redding, CA).
Grids were heated to 95C for 10 min, and resin was etched from the sections as described in Groos et al. (2001). Etched sections were rinsed in 0.05 M HEPES buffer for 5 min, followed by incubation in 0.05 M glycine in HEPES to neutralize reactive aldehyde groups. Nonspecific binding sites were blocked with 5% BSA in HEPES for 15 min. Grids with ultrathin sections of skeletal muscle tissue or sections of human platelets were incubated with a mixture of primary conjugates as mouse anti-myosin antibodies conjugated to cAu6 and rabbit anti-α-actinin IgG conjugated to cPd6 for skeletal muscle samples or with mouse anti-actin cPd6 and rabbit anti-α-actinin cAu6 conjugates in case of platelet samples for 1 hr at room temperature. Primary conjugates were used at a concentration of 1 × 1013 particles/ml (Park et al. 1987).
After washing five times with PBS for 2 min each, sections were further incubated with a mixture of secondary IgG conjugated to fluorophores, donkey anti-mouse IgG conjugated to Cy3, and donkey anti-rabbit antibodies conjugated to Cy2 (Jackson ImmunoResearch; Westgrove, PA).
Secondary fluorophore conjugates were diluted 1:80 in PBS, pH 7.4. After rinsing with PBS five times for 2 min each, samples were transferred into small Petri dishes with cover-glasses at the bottom (Mattek Cultureware; Ashland, MA).
Fluorescence Light Microscopy
Samples were observed with a Zeiss Axiovert 200M microscope, and images were taken with an Axiocam HRc (Zeiss; Oberkochen, Germany) camera. Filters were set at Cy2- or Cy3-specific emission (em)/excitation (ex) wavelength, for Cy2 (ex: 470 ± 20 nm; em: 540 ± 15 nm) or Cy3 (ex: 546 ± 6 nm; em: long-pass filter > .590 nm) filters. During observation, grids were submerged in PBS. After LM, grids were fixed in 0.1% glutaraldehyde in PBS, pH 7.4, for 15 min, rinsed briefly twice in ddH2O, and air dried. For LM controls, primary antibody conjugates were omitted, and samples were incubated with secondary antibody Cy2 and Cy3 conjugates only. Grids were thin coated with carbon to increase the stability of sections during imaging.
TEM and EFTEM
Specimens were examined with a Zeiss EFTEM 912 with a LaB6 filament and an emission current of 12 μA with 120 kV acceleration voltage. Gold and palladium particles were identified with ESI applying the three-window method at energy losses of 75, 45, and 35 eV for the AuO2,3-ionization edge and 430, 324, and 260 eV for the PdM4,5-edge. For detection of cAu6, exposure time was 2 sec at an illumination angle of 1.25 mrad. Exposure time for detection of cPd6 was increased up to 15 sec at an illumination angle of 3.15 mrad because signal intensities decrease with higher energy losses. Energy-filtered images were analyzed by the power law method using EsiVision software from SIS, and images of 1024 × 1024 pixels were taken with a slow scan CCD camera (SIS; Muenster, Germany).
Results
Skeletal Muscle Model
Ultrathin Epon sections of rat skeletal muscle tissue were etched and labeled with primary mouse anti-myosin cAu6 and goat anti-α-actinin cPd6primary conjugates for EM. For LM, the specimen-adherent primary bound anti-myosin anti-cAu6. Anti-α-actinin cPd6 conjugates were labeled with Cy3- and Cy2-coupled secondary donkey anti-mouse and donkey anti-goat antibodies, respectively. Cy3 anti-myosin signal appeared as a broad-banded regular pattern, whereas Cy2, α-actinin, fluorescence was restricted to narrow bands (Figure 1). Observation of the exact same sample area by TEM and analyses of the metal labels by EFTEM confirmed the presence of Au over myosin-containing A bands and distribution of Pd over α-actinin-containing Z lines (Figure 2).
Platelet Model
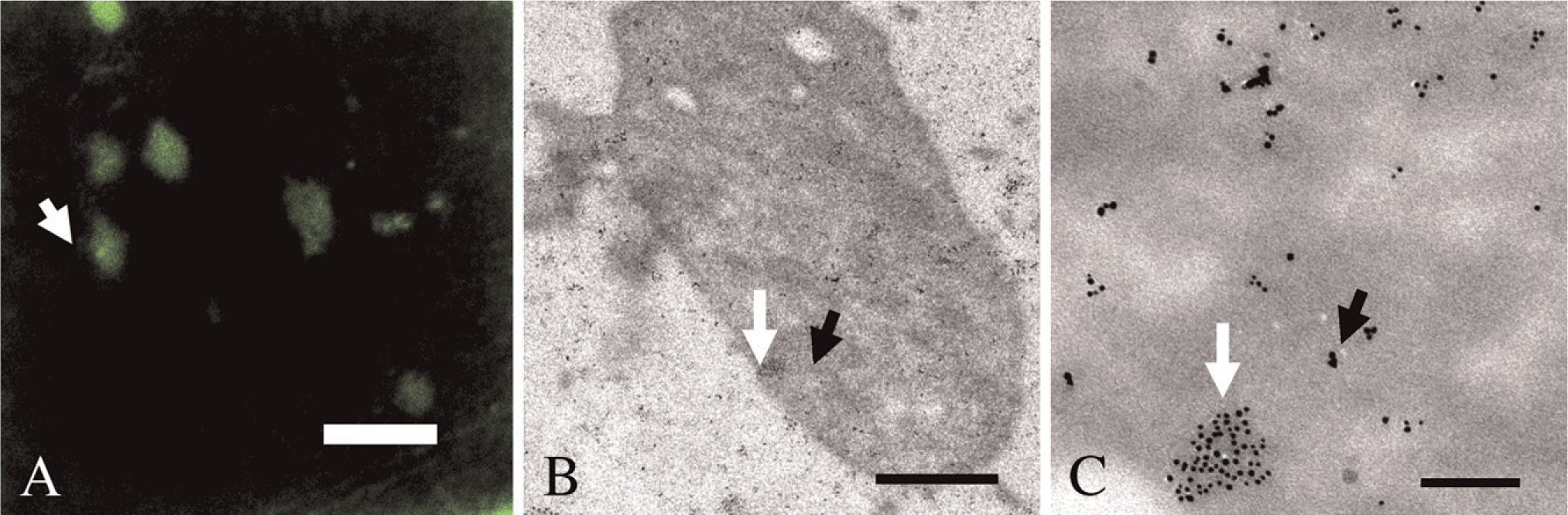
Sections of human platelets embedded in Epon were etched and incubated with mouse anti-actin cPd6 and rabbit anti-α-actinin cAu6 primary antibodies. Primary antibody conjugates were detected for LM with donkey anti-mouse Cy3 and donkey anti-rabbit Cy2 secondary fluorophore–antibody complexes (Figure 3). Anti-α-actinin signal was concentrated mainly in bright, clearly defined foci and distributed in the cytoplasm of platelets with lower intensity. In contrast, actin signal was exclusively present in a punctate pattern within the cytoplasm of platelets. Foci that were bright with anti-α-actinin signal were not labeled by anti-actin-specific signal. In EM, the same labeling patterns as for LM were observed (Figure 4).
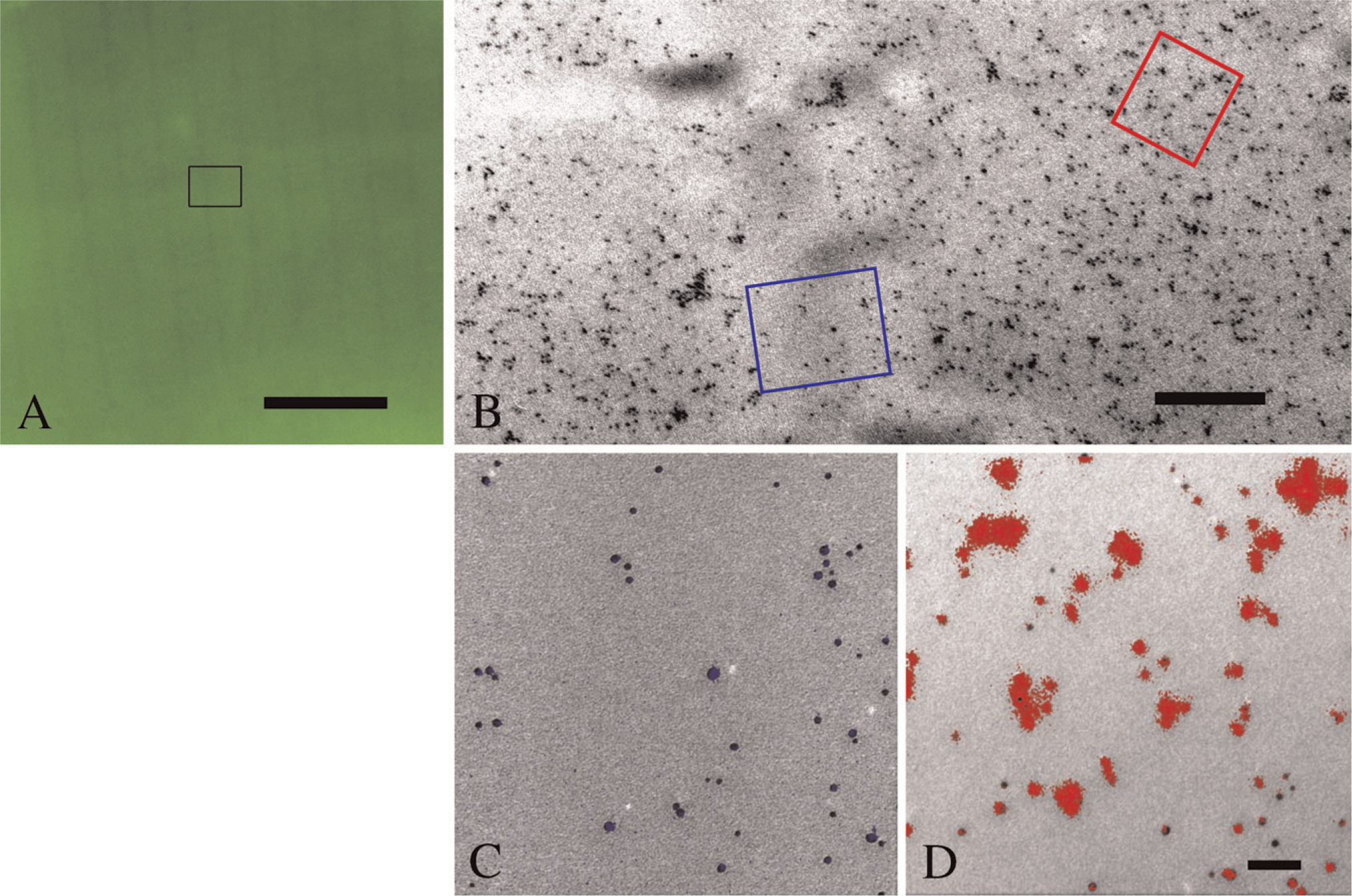
Elemental analysis by ESI of granules showing strong α-actinin-specific fluorescence in LM revealed accumulation of cAu corresponding to anti-α-actinin cAu6 conjugates (Figures 5A–5D). In the same area, the weaker, diffuse α-actinin-specific fluorescent signal was observed in the cytoplasm of platelets with LM, the particle demonstrating a palladium-specific signal representing cPd6 anti-actin conjugates when analyzed with ESI (Figures 5E–5H). Adjacent colloidal gold and colloidal palladium nanoparticles could be differentiated using either gold ESI or the palladium ESI data (Figures 5C, 5D, 5G, and 5H).
Discussion
Correlative immunolabeling permits the acquisition of multiple data sets from a single sample by more than one microscopic technique (Takizawa et al. 1998; Robinson et al. 2001). To date, studies have often utilized serial resin sections for LM and EM (Groos et al. 2001). Because separate sections are used for observation at LM and EM, it is not always possible to extract direct information from the same part of the tissue with the two different microscopy techniques (Sun et al. 1995; Richter et al. 2005). In this study we have simultaneously labeled two antigens such that the exact same-labeled sites on a section can be observed consecutively by LM and EM. This allows rapid evaluation of labeling reactions via LM and mapping areas of interest at the cellular level for further investigation with EM at higher levels of spatial resolution. Identification of specific association with cellular structural elements or interactions of different molecular species in macromolecular complexes can be addressed using this technology.

Light micrographs of 60-nm rat skeletal muscle tissue section. Samples were embedded in Epon, sectioned, and incubated with two different labels. The first label is mouse anti-myosin IgG conjugated to 6-nm colloidal gold nanoparticles, cAu6, and labeled with secondary antibody conjugated to Cy2. The second label is goat anti-α-actinin IgG conjugated to 6-nm colloidal palladium nanoparticles, cPd6, and labeled by secondary antibody conjugated to Cy3. In the same area,
When colloidal metal markers of different sizes are used to differentiate epitopes, the metal markers need to differ considerably in nominal diameter to avoid overlap due to variation around “nominal” particle sizes. Markers with different sizes show different labeling efficiencies because larger labels can bind to, block, or mask more binding sites per marker compared with smaller labels (Horisberger 1981). Therefore, differences in labeling efficiencies caused by different-sized cAu particles can be eliminated by the use of equal-sized particles of different elemental composition that can be identified by ESI (Meyer and Albrecht 2001; Kandela et al. 2005). Also, the spatial resolution and precision of localization attainable in EM can be impaired by attaching colloidal metal particles to secondary or tertiary antibodies, which place the particles at a distance from epitopes. Conjugating colloidal metal particles to primary antibodies provides the highest spatial resolution attainable in EM. Lower resolution of LM is not affected by larger distance of fluorophores bound to secondary antibodies from antigenic binding sites. It is possible to synthesize particles with a 5- to 10-nm range in diameter with small variation in size such that particles with a 2- to 3-nm difference in nominal size can be reliably identified. Here several different particle sizes could be used relatively effectively in the 3- to 10-nm range (Meyer and Albrecht 2003). However, we find the use to be impractical timewise because each label on a specimen has to be accurately measured to determine its exact size in order to unambiguously identify the label and epitope.
An ideal label for correlative immunolabeling would consist of both reporters, for LM as well as EM, bound to the same antibody or ligand molecule. However, when cAu markers and fluorophores are in close vicinity, the fluorescent signal is quenched by resonant energy transfer (Powell et al. 1997; Dulkeith et al. 2002). Our studies have shown that at close range, when colloidal metal particles and fluorophores were conjugated to primary antibodies, the degree of quenching was virtually 100% and did not depend on the diameter of colloidal metal particles (Kandela et al. 2004; Kandela and Albrecht in press). By increasing the distance between the fluorophores and colloidal metal particles, quenching is reduced. However, in this case the degree of quenching is influenced both by the size of the metal particles and the distance. Larger sizes of metal particles show greater quenching when the fluorophore is conjugated to the secondary antibody (Kandela et al. 2003). Approximately 50% of the fluorescent signal from the secondary antibody remained when cAu particles with a 6-nm diameter were attached to primary antibodies and ∼20% remained if the primary antibody was conjugated to 18-nm particles.
Electron micrographs of rat skeletal muscle tissue. Sample was embedded in Epon. Sections were labeled with cPd conjugated to anti-α-actinin and cAu conjugated to anti-myosin. (
In the present study we used primary antibodies conjugated to colloidal metal particles (cAu6 and cPd6) as reporters for EM and fluorophores attached to secondary antibodies (Cy2 and Cy3) as reporters for LM. The system insures a high spatial resolution for EM without compromising LM resolution (Ahn and Krivanek 1983; Nisman et al. 2004).
Multiple correlative immunolabeling for LM and EM was used here on two model systems. α-Actinin and myosin were localized on resin sections of skeletal muscle tissue samples, and α-actinin and actin were labeled on sections of embedded platelets. Labeling patterns corresponded to the known distribution of the antigens in both samples (Dubernard et al. 1997), and the fluorescent signals observed by LM were identical to the positions of respective metal labels when the same sample areas were subsequently observed with EM. Signal intensities correlated well between LM and EM: in labeled muscle samples with uniform fluorescent intensities, metal markers were distributed over the entire labeled area, whereas in platelet samples bright fluorescent spots represented areas covered by more dense focal accumulation of metal labels.

Light micrographs of Epon-embedded platelets. Sections are 60-nm thick. Platelets were labeled for actin and α-actinin. (
Conjugation of luminescent semiconductor quantum dots to antibodies is also a promising technology for correlative labeling. The particles are fluorescent and sufficiently electron dense to be observed in EM (Alivisatos et al. 2005; Deerinck et al. 2005) Currently, however, although the “quantum” portion of the dot is small (5–10 nm), the stabilizing and functional coatings substantially enlarge the particles. We have found that particle size variation is also significant. For simultaneous multiple labeling, the quantum core can be synthesized of different elemental compositions. However, their small size and elemental composition can make identification via EFTEM difficult, although recent studies along these lines have been encouraging (Nisman et al. 2004).
Very small clusters of gold atoms conjugated to fluorescent molecules or “fluoronanoprobes” do not show quenching of the fluorescent signal. These are useful in the LM and EM correlation of individual antigenic species. However, due to the very small amount of metal, for most practical purposes the probes require enhancement by additional gold or silver. (Takizawa et al. 1998; Robinson et al. 2000).
Correlative double immunolabeling of an Epon section of resting, inactivated human platelets. Anti-α-actinin-specific fluorescence signal from Cy2 (
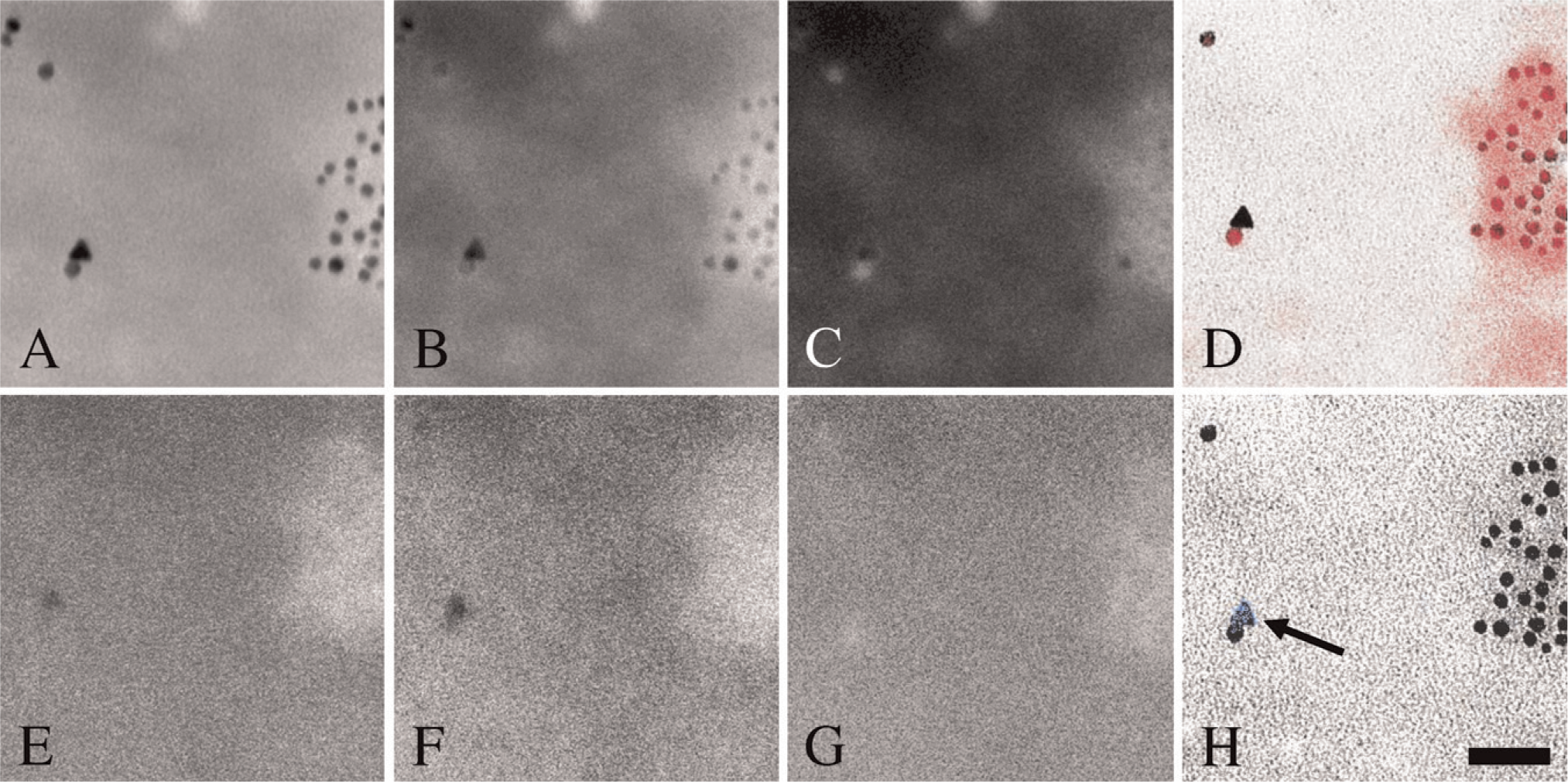
ESI analysis of markers over white arrowhead in Figure 4. Background images taken below Au-specific (
The technology reported here provides a practical method to combine LM and EM with correlative multiple immunolabeling using different fluorophores and colloidal metal nanoparticles as labels. The method limits quenching of fluorescent dyes for LM and provides molecular spatial resolution for EM of the exact same labels. Simultaneous multiple correlative immunolabeling to detect more than two antigens can be achieved using metal nanoparticles of different compositions such as Au, Ag, Pd, Pt, or Fe on primary antibody of antigenically distinct origin and appropriate fluorescent dyes coupled to secondary antibodies, specific for the primary antibodies.
EFTEM was useful in the differentiation of the various small colloidal particles. The application of energy-dispersive x-ray (EDX) analysis would be useful in that scanning TEM (STEM) and SEM as well as TEM could be used and small particle labels in thick sections or whole-mount preparations could potentially be analyzed. Based on preliminary studies using an aberration corrected field emission STEM with a very stable stage and high-resolution EDX detector, we feel the 3-nm and larger size range is possible.
Footnotes
Acknowledgements
This study was supported by the National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS) Grants #63001 and #67244.