
Abstract
Transglutaminases (TGs) are protein crosslinking enzymes involved in cell adhesion and signaling and matrix stabilization and maturation, in many cell types and tissues. We previously described that in addition to transglutaminase 2 (TG2), cultured MC3T3-E1 osteoblasts also express the plasma TG Factor XIIIA (FXIIIA). Here we report on the expression and localization of FXIIIA in bone in vivo and provide confirmatory in vitro data. Immuno-histochemistry and in situ hybridization demonstrated that FXIIIA is expressed by osteoblasts and osteocytes in long bones formed by endochondral ossification (femur) and flat bones formed primarily by intramembranous ossification (calvaria and mandible). FXIIIA immuno-reactivity was localized to osteoblasts, osteocytes, and the osteoid. RT-PCR analysis revealed FXIIIA expression by both primary osteoblasts and by the MC3T3-E1 osteoblast cell line. Western blot analysis of bone and MC3T3-E1 culture extracts demonstrated that FXIIIA is produced mainly as a small, 37-kDa form. Sequential RT-PCR analysis using overlapping PCR primers spanning the full FXIIIA gene showed that the entire FXIIIA gene is expressed, thus indicating that the 37-kDa FXIIIA is not a splice variant but a product of posttranslational proteolytic processing. Forskolin inhibition of osteoblast differentiation revealed that FXIIIA processing is regulated by the protein kinase A pathway.
E
Transglutaminase (TG) enzymes have been implicated in FN and COL I matrix stabilization in many tissues including bone (Mosher and Schad 1979; McDonald et al. 1982; Barry and Mosher 1988; Al-Jallad et al. 2006). TGs are a family of widely distributed enzymes (currently nine genes are known (TG1, TG2, TG3, TG4, TG5, TG6, TG7, Factor XIIIA, and inactive erythrocyte TG) that catalyze a Ca2+-dependent acyl-transfer reaction between polypeptide-bound glutamine residues and primary amines (Grenard et al. 2001; Lorand and Graham 2003). The amines, predominantly acting as acyl acceptors, are protein-bound lysine residues and polyamines such as putrescine and spermidine (Lorand and Graham 2003). This enzymatic reaction is exclusively performed by TGs and can take place in the cytosol, at the cell surface, and/or in ECM compartments (Lorand and Graham 2003). The resulting covalent γ-(glutamyl)-ε-lysyl bond (an isopeptide crosslink) can form intra- or intermolecularly within or between substrate proteins (Folk and Cole 1966; Green-berg et al. 1991; Aeschlimann and Thomazy 2000). The isopeptide crosslinks are resistant to normal proteolysis and thus are thought to enhance biochemical stability and cohesive integrity of ECM in normal tissues and in wound healing and scarring (Aeschlimann and Thomazy 2000; Verderio et al. 2005).
Of the nine currently known TG enzymes, only TG2 and Factor XIIIA (FXIIIA) are associated with formation of skeletal elements where published reports have focused on cartilage and endochondral ossification (Aeschlimann et al. 1993, 1996; Nurminskaya et al. 1998; Kaartinen et al. 2002, 2005; Nurminskaya and Linsenmayer 2002; Nurminskaya and Kaartinen 2006). TG2 is localized in vivo to hypertrophic chondrocytes (Aeschlimann et al. 1993, 1996), osteoblasts, osteocytes, and odontoblasts (Kaartinen et al. 2002, 2005). In cartilage, both TG2 and FXIIIA expression are upregulated in hypertrophic chondrocytes in the growth plate during embryonic limb development (Aeschlimann et al. 1993, 1996; Nurminskaya et al. 1998; Nurminskaya and Linsenmayer 2002), and extracellular TG2-GTP (guanosine triphosphate) complexes have been shown to promote chondrocyte hypertrophy and cartilage mineralization (Johnson and Terkeltaub 2005). Moreover, chondrocyte-derived TGs promote mineralization of chondrocyte-osteoblast cocultures (Nurminskaya et al. 2003). We have recently demonstrated in vitro that abundant TG-mediated matrix crosslinking occurs during cell differentiation and ECM assembly in the mouse calvarial MC3T3-E1 osteoblast cell line; TG genes expressed and active in the cultures were TG2 and FXIIIA (Al-Jallad et al. 2006). Moreover, inhibition of matrix crosslinking in these cultures by a TG inhibitor resulted in a dramatic downregulation of FN and COL I expression, which in turn blocked osteoblast differentiation and mineralization of the cultures. Collectively, these findings indicate that TG activity is critical for osteoblast differentiation and matrix maturation (Al-Jallad et al. 2006).
TG2 was the first TG enzyme described in mineralized tissues (growth plate cartilage and bone), but another TG was thought to exist (Aeschlimann et al. 1993, 1996; Kaartinen et al. 2002). Further work on cartilage by Nurminskaya et al. (1998) and Nurminskaya and Linsenmayer (2002) showed that this TG is FXIIIA, an observation recently extended by us toward other mineralized tissues through data obtained from an osteoblast cell culture model (Al-Jallad et al. 2006). In further support of there being at least a second TG protein providing TG crosslinking activity influencing cell differentiation and matrix assembly and mineralization in chondrocyte and osteoblast cultures is the observation that TG2-knockout mice (Tgm2−/-) have no overt skeletal phenotype (De Laurenzi and Melino 2001; Nanda et al. 2001). Perhaps most significant in this regard was our preliminary analysis of bones and teeth from Tgm2−/- mice showing that, even in the absence of TG2, TG enzymatic activity levels were similar to their wild-type counterparts (Nurminskaya and Kaartinen 2006). Moreover, no changes in polymerization (cross-linking) levels of a major bone substrate protein (osteopontin) were observed (Nurminskaya and Kaartinen 2006), demonstrating that crosslinking is nevertheless taking place in the bones of Tgm2-deficient mice, and that at least some of the substrate proteins are the same, indicating at least partially overlapping functions for TG2 and a second TG in bone. Also consistent with the presence of another TG in bone was our observation that thrombin treatment (which activates FXIIIA) of both the wild-type and Tgm2−/- knockout bone extracts increased TG activity in the extracts, thus implicating enzymatic activity derived from FXIIIA (Nurminskaya and Kaartinen 2006).
In this study, to further explore the possibility that FXIIIA is an enzyme active in bone with important physiological functions, we show by immunohistochemistry (IHC), in situ hybridization, and biochemical methods that FXIIIA is expressed in vivo by osteoblasts and osteocytes in bones formed by both intramembranous and endochondral ossification. Moreover, we report that FXIIIA is present in both bone tissue and in osteoblast cultures mostly as a small, 37-kDa form, likely resulting from posttranslational proteolytic processing of the parent enzyme. The processing is controlled by protein kinase A and appears to define FXIIIA localization to the osteoblast cell surface. This 37-kDa FXIIIA was also found in chondrocytes and macrophages, but not in plasma, indicating that it might represent the cell and tissue form of FXIIIA.
Materials and Methods
Tissue Preparation for Light Microscopy
Four-month-old Balb/c mice were sacrificed by cervical dislocation, and the mandibles, calvariae, and femora were immediately dissected and immersed in fixative (4% paraformaldehyde in 0.1 M sodium cacodylate buffer, pH 7.4, for 1 day at 4C). Bones were decalcified in 10% EDTA at 4C for 10 days and then dehydrated through a graded series of ethanol followed by routine embedding in paraffin. Paraffin sections for histological analysis and for IHC were prepared on a rotary microtome. All animal experiment protocols were approved by the Animal Care Committee of McGill University (Montreal, Canada) and the M.D. Anderson Cancer Center (Houston, TX).
IHC for FXIIIA
For light microscopic immunolocalization of FXIIIA, paraffin sections of mouse bones were deparaffinized and incubated with 2.5 mM trypsin in 5 mM Tris-HCl buffer (pH 7.3) supplemented with 2.25 mM CaCl2 (Sigma; St Louis, MO) for 20 min at 37C. Following a blocking treatment with PBS containing 10% swine and 5% goat sera, sections were incubated with rabbit anti-human FXIIIA antibody (ab717; Abcam, Cambridge, MA) for 12 hr at room temperature. A biotinylated swine anti-rabbit IgG F(ab')2 fraction (Dako Cytomation; Carpinteria, CA) was used as the secondary antibody and incubated with the section for 1 hr at room temperature. For FXIIIA IHC, mouse anti-human FXIIIA primary antibody (AC-1A1; Labvision, Fremont, CA) incubation was followed by blocking and secondary antibody treatments using HISTOMOUSE-SP KIT (Zymed Laboratories, Invitrogen Immunodetection; South San Francisco, CA) according to manufacturer's instructions. Following incubation with EzLink Extra Avidin-Alkaline Phosphatase (Sigma) for 30 min, immunoreactivity was visualized using a Vector Red kit (Vector Laboratories; Burlingame, CA) resulting in pink/red color for positive staining. Counterstaining was performed with either methyl green or hematoxylin. Normal rabbit sera was used as a replacement for primary antibody in negative-control incubations.
In Situ Hybridization for FXIIIA
Following deparaffinization, sections were treated with 0.2 M HCl for 20 min at room temperature and digested with 5 μg/ml proteinase K at 37C for 15 min. Sections were then postfixed with 4% paraformaldehyde/PBS solution, immersed in 2 mg/ml glycine/PBS for 30 min, and kept in 40% deionized formamide in 4X SSC (standard saline citrate) until hybridization. Hybridization was carried out at 37C for 15 hr with FITC-conjugated oligo-cDNA (60mer) for the mouse FXIIIA gene (F13a1) (GenBank AK036403) (GeneDetect.com Ltd; Bradenton, FL). After a series of rinses with 2X SSC, reaction sites were visualized by GenPoint Fluorescein kit (Dako) according to the manufacturer's instructions. Brown color represented positive staining, and methyl green was used for counterstaining. Sense probe was used as the negative control following the same methodological protocol.
Osteoblast Cultures
MC3T3-E1 (subclone C14) preosteoblast cells were a generous gift from Dr. Renny T. Franceschi (University of Michigan School of Medicine; Ann Arbor, MI). Cells were routinely cultured in modified alpha minimum essential medium lacking ascorbic acid (AA) (Invitrogen; Carlsbad, CA) supplemented with 10% FBS (Hyclone; South Logan, UT) and 1% penicillin-streptomycin (Invitrogen). To differentiate the cells and mineralize the cultures, cells were grown for 12 days in the presence of 50 μg/ml AA (a critical cofactor required for collagen production) and 10 mM β-glycerophosphate (βGP; an organic source of phosphate required for mineralization) in a humidified 5% CO2 incubator at 37C as described previously (Al-Jallad et al. 2006). Where described, cells were also treated with 10 μM forskolin (EMD Biosciences; Darmstadt, Germany) to inhibit osteoblast differentiation and in vitro bone formation. Forskolin was not toxic to the cells at 10 μM concentration tested by MTT (3-[4,5-dimethyl-thiazol-2-yl]-2,5-diphenyltetrazolium bromide; thiazolyl blue) incorporation to viable cells (Sigma) according to the manufacturer's instructions. After 12 days of culture, cells were processed for von Kossa staining to visualize mineralization and for Picrosirius staining to visualize and quantify collagen deposition. Both methods were carried out as described previously (Al-Jallad et al. 2006). For protein analysis, cell layers (including abundant mineralized ECM) were harvested with RIPA buffer containing 5 mM EDTA and 5 mM protease inhibitor cocktail (Sigma). Cell extracts were homogenized by ultrasonication and then centrifuged. The supernatant obtained from this protocol was used as the protein extract. Protein concentration was determined using the bichinchonic acid (BCA) protein assay kit (Pierce Biotechnology; Rockford, IL). Subcellular protein fractionation of the cells was done using ProteoExtract Subcellular Proteome Extraction Kit (EMD Biosciences) according to the manufacturer's instructions.
Bone Extracts
Tibias and femurs were excised from 1-month-old wild-type and Tgm2−/- knockout mice immediately after killing by cervical dislocation. Epiphyseal heads including the growth plates and parts of the metaphyses were removed from the two ends of the femurs to exclude cartilage from the analyses. Rat bones were processed as previously described (Kaartinen et al. 2002, 2005). Tissue extracts were prepared as previously described (Goldberg et al. 1988; Greenberg et al. 1991; Kaartinen et al. 2002, 2005) by sequential extractions with 4 M guanidium-HCl (G1 extract), 0.5 M EDTA (E extract), and 4 M guanidium-HCl (G2 extract). This sequential extraction scheme allows discrimination between mineral-binding proteins and proteins from non-mineralized cell and tissue compartments. For TG activity assays, the G1 extract was re-extracted with 10 mM Tris buffer (pH 7.4) containing 0.2% IGEPAL and 2 mM PMSF (G1/T extract) using MICROCON YM-30 concentrators (Millipore; Billerica, MA).
RT-PCR
Total RNA was isolated from the mineralized MC3T3-E1 osteoblast cell cultures using Trizol reagent (Invitrogen). RNA extract of mouse primary osteoblasts was a generous gift from Dr. René St-Arnaud (Shriners Hospital; and Department of Human Genetics, Faculty of Medicine, McGill University, Montreal, Canada). One μg total RNA was reverse transcribed and amplified using Superscript one-step RT-PCR with Platinum Taq (Invitrogen). All primer sequences and PCR conditions are listed in Table 1. Amplified products were analyzed by 2% agarose gel electrophoresis and stained with ethidium bromide.
SDS-PAGE and Western Blotting
Protein samples, 20 μg each, from G1, E, and G2 extracts from wild-type and Tgm2−/- mouse bones, MC3T3-E1 osteoblast cell extracts, human plasma (Aniara Corporation; Mason, OH), purified plasma FXIII (EMD Biosciences), mouse embryonic chondrocyte extract (hypertrophic chondrocytes from the E18 tissues, kindly donated by Dr. Maria Nurminskaya; Department of Anatomy and Cell Biology, Tufts University, Boston, MA), and mouse macrophage extract (Becton Dickinson; Franklin Lakes, NJ) were separated on 10% SDS-acrylamide gels under reducing conditions and subsequently transferred onto PVDF membranes (BioRad Laboratories; Hercules, CA) for Western blotting or processed for silver staining (Blum et al. 1987). For Western blotting, membranes were blocked with 5% skim milk overnight at room temperature and then incubated with mouse anti-FXIIIA monoclonal antibody (AC-1A1; Labvision). Horseradish peroxidase (HRP)-conjugated sheep anti-mouse IgG (Amersham Bioscience; Piscataway, NJ) was used as a secondary antibody for a 1-hr incubation at room temperature. Reactions were visualized using an ECL Plus kit (Amer-sham Bioscience). Where indicated in the figures, samples were incubated with 0.76 μg/ml thrombin (Sigma) for 30 min at 37C in Tris-buffered saline (TBS) containing 3 mM CaCl2, at pH 6.8, prior to running SDS-PAGE. Purified human FXIIIA (EMD Biosciences) and osteopontin were used as positive controls for thrombin cleavage.
RT-PCR primers used in FXIIIA analysis
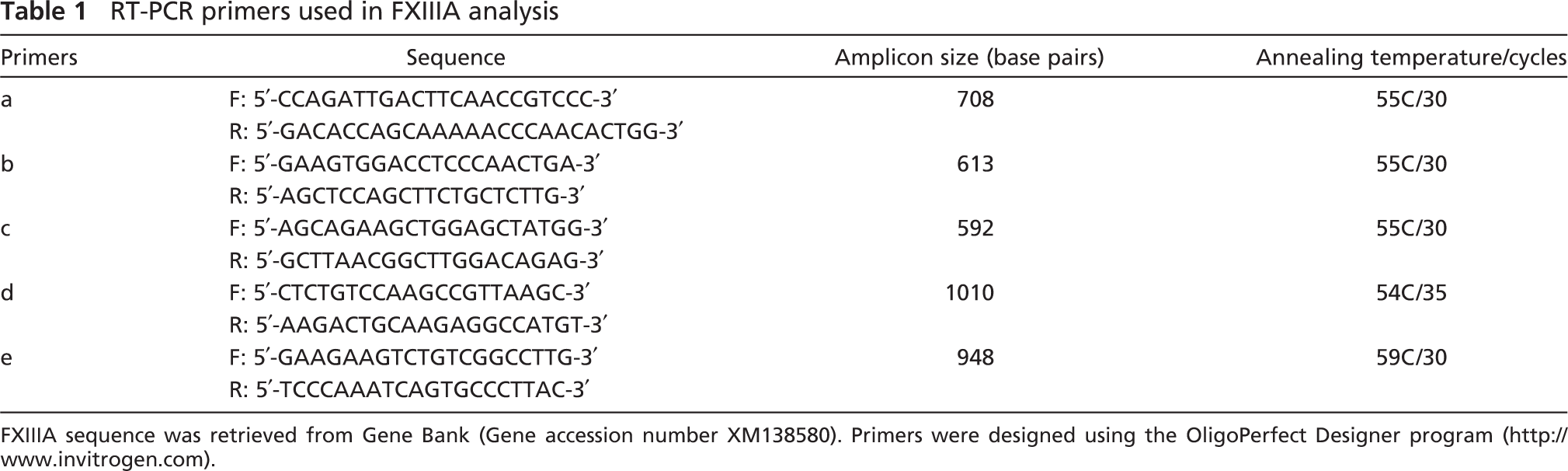
FXIIIA sequence was retrieved from Gene Bank (Gene accession number XM138580). Primers were designed using the OligoPerfect Designer program (http://www.invitrogen.com).
TG Activity Assays
G1/T extract (12 μg) was treated with 0.76 μg/ml thrombin as described above and further incubated with 5 mM 5(biotin)-pentylamine (bPA;Pierce Biotechnology), 1 mM dithiothreitol, and 1 μg FN (Sigma) for 2 hr at 37C. Reaction products were separated by 10% SDS-PAGE and blotted onto PVDF membranes. Membranes were blocked with 5% non-fat milk followed by incubation with EzLink Extra Avidin-HRP (Sigma) for 2 hr at room temperature. Immunoreactive bands were visualized using an ECL Plus kit (Amersham Bioscience).
Results
IHC Localization of FXIIIA in Bone
The presence of FXIIIA in bone was examined by IHC in long bone (femur) and in flat bones (calvaria and mandibular alveolar bone) using rabbit polyclonal antibody (ab717) raised against human FXIIIA and using mouse anti-human monoclonal antibody (AC-1A1). In all bone types examined by IHC, cellular FXIIIA immuno-reactions were found in osteoblasts and osteocytes (Figures 1A-1C). In the ECM, weak staining was also observed in the osteoid, particularly in long bones. Negative controls without primary antibody did not show any reaction (data not shown). Identical immunolocalization patterns were obtained with both antibodies (data shown for ab717).
Expression of FXIIIA mRNA in Bone Cells
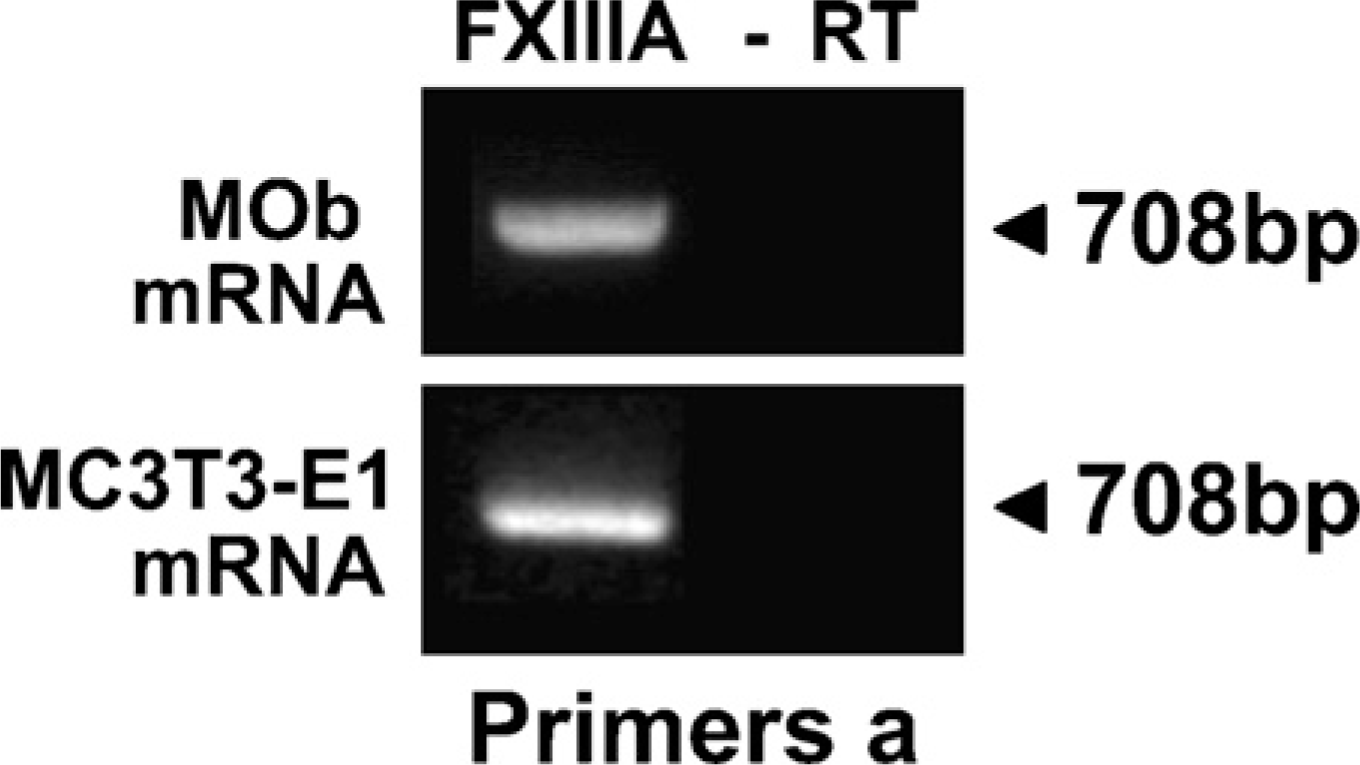
In situ hybridization signals for FXIIIA mRNA were observed in the cytoplasm of osteoblasts and osteocytes in all types of bones analyzed (Figures 1D-1F). No signal was detected by the sense probe. Similarly, RT-PCR analysis of the RNA extract from both mouse calvarial primary osteoblasts and MC3T3-E1 osteoblasts revealed the presence of mRNA for FXIIIA (Figure 2).
FXIIIA Protein Expression
The presence of FXIIIA in bone was also examined by Western blotting. To confirm the specificity of the monoclonal antibody (AC-1A1) used for this detection, the antibody was tested against purified human plasma FXIII and complete human plasma. As shown in Figures 3A and 3B, the antibody detected only FXIIIA and not FXIIIB. The antibody also detected FXIIIA before and after thrombin activation (FXIIIa) (Figures 3A and 3B). Specificity of the antibody was further tested against complete human plasma (after albumin removal) where the antibody detected FXIIIA at ~80 kDa and 55 kDa; the faint band observed at 69 kDa is albumin (Figure 3C).
Long bone (femurs and tibias after removal of marrow and growth plates) G1, E, and G2 extracts were used to demonstrate FXIIIA protein in bone. The G1 extract generally contains organic constituents from all unmineralized tissue compartments in the bone such as the cells, the osteoid layer, and other connective tissue components of bone. The E extract contains mineral-binding proteins, and the G2 extract contains additional ECM non-collagenous proteins and soluble collagen (Fisher et al. 1987; Goldberg et al. 1988). By Western blotting, the majority of FXIIIA protein was detected at 37 kDa. Weak 80 kDa and 75 kDa bands, which likely represent the full-length unactivated form and the activated form, respectively, were also seen. A faint 55-kDa FXIIIA band was also present. Because we have shown that osteoblast TG activity arises from two TGs—TG2 and FXIIIA—and have speculated that overlapping functions might explain the lack of a bone phenotype in Tgm2−/- mice, we analyzed FXIIIA protein levels also in Tgm2−/- bone extracts. As demonstrated in Figure 4A, FXIIIA protein levels in both wild-type and Tgm2−/- bones are similar, and no substantial overexpression of FXIIIA was observed in the knockout bones. In both wild-type and Tgm2−/- bone preparations, FXIIIA was found predominantly in the G1 and E extracts, with the G1 extract clearly having the largest fraction of the protein (Figure 4A). Because the G1 extract generally contains the proteins of bone cells and of the osteoid layer, Western blot data are consistent with the in situ hybridization and IHC data showing the location of message and protein, respectively, in osteoblasts, osteocytes, and osteoid. Collectively, these similar in vitro and in vivo observations indicate that the FXIIIA found in bone extracts likely derives from bone cells and not from other connective tissue components such as blood vessels.
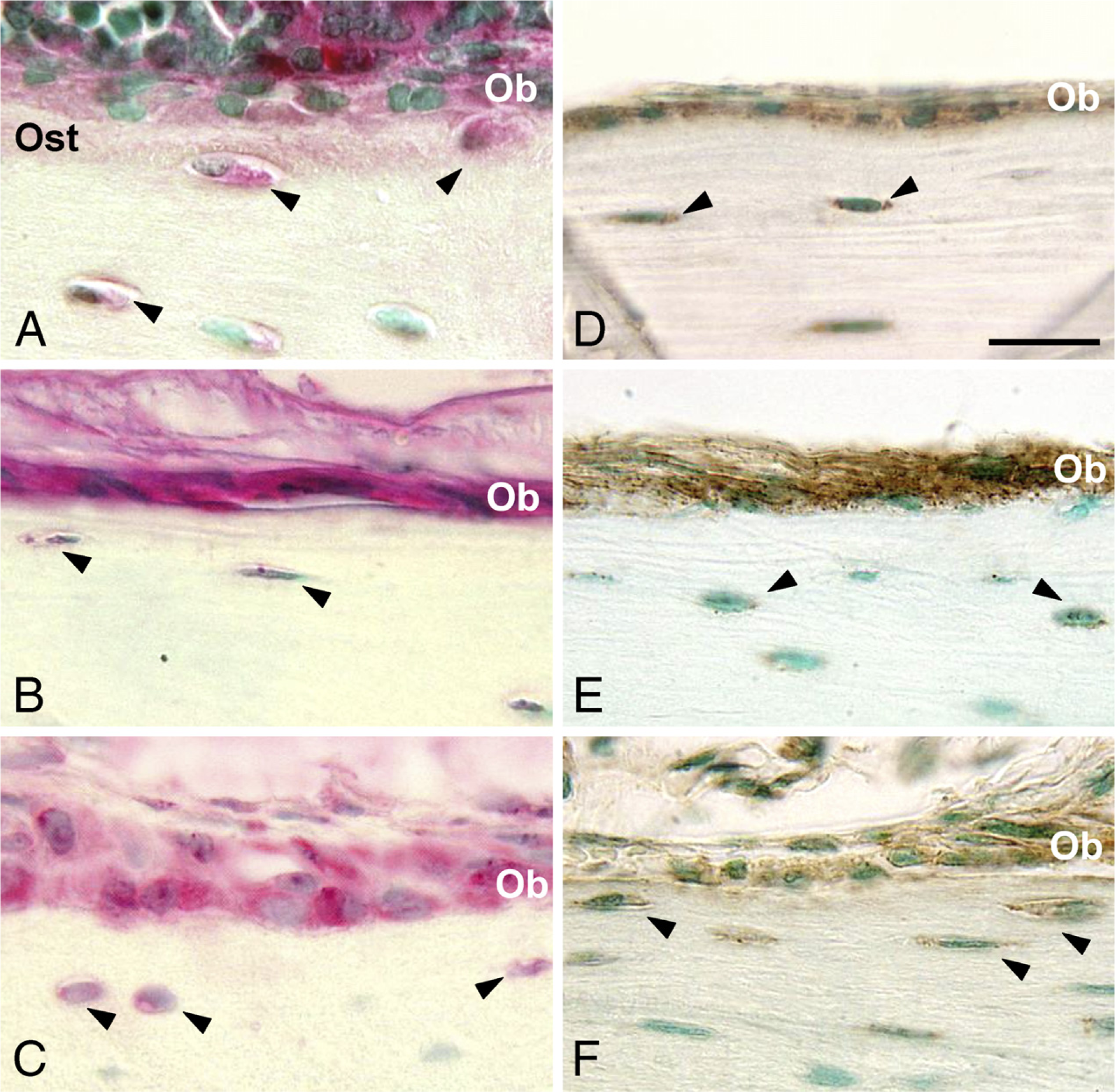
Factor XIIIA (FXIIIA) protein and mRNA expression in bone. (
Analysis of FXIIIA expression in MC3T3-E1 osteoblast cultures revealed a strong detection of 37-kDa FXIIIA (Figure 4B). Because this form was not found in plasma, we hypothesized that it might represent a cellular or tissue form of FXIIIA. To test this hypothesis, we analyzed mouse chondrocyte extracts and macrophage cell extracts, both reported to contain abundant FXIIIA (Nurminskaya and Linsenmayer 2002; Adany and Bardos 2003). The extracts from both these cell types similarly demonstrated the small 37-kDa form of FXIIIA (Figure 4B), where it was the most prominent band in the preparations. To address a question whether the form is species specific and only found in mouse cells and tissues, we analyzed rat bone and found the same 37-kDa form (Figure 4C).
TG Activity in Bone Extracts
Because we have previously shown that TG activity is found in bone E extracts (Kaartinen et al. 2002; Nurminskaya and Kaartinen 2006), we sought to determine whether the G1 extract, where the majority of the FXIIIA is found, also contains some level of TG activity. To analyze G1 extracts that are normally soluble in denaturing 8 M urea, we re-extracted the material with Tris-buffer (G1/T extract). Results from the TG-activity assay from wild-type and Tgm2−/- mouse bones show clear TG enzymatic activity as evidenced by primary amine incorporation into the FN substrate (Figure 5A). Although the activity in the G1/T extract appears slightly higher in wild-type bones as compared with bones from Tgm2−/- mice, the values were not statistically significant. Importantly, the observed activity was not increased by thrombin, implying that most of the FXIIIA present in the tissue (37-kDa form) did not require activation or was thrombin insensitive. Cleavage experiments using bone and cell extracts showed that thrombin does not appear to cleave the 37-kDa FXIIIA form (Figure 5B); however, cleavage products <1 kDa are difficult to visualize by Western blotting. Activity of thrombin under these conditions was confirmed by observing successful cleavage of full-length FXIIIA and osteopontin (data not shown).
Proteolytic Processing of FXIIIA During Osteoblast Differentiation
To investigate whether the 37-kDa form of FXIIIA in osteoblasts derives from one transcript or perhaps arises from separate products of smaller mRNA splice variants, we designed primers spanning the entire gene to examine the expression of all FXIIIA exons (Table 1) in the MC3T3-E1 osteoblast cells. Our data demonstrate that all exons are transcribed and that there are no smaller variant mRNAs produced (Figure 6), indicating that the form of FXIIIA that we have observed is a result of posttranscriptional proteolytic prosessing of this protein. To gain some understanding of the role of the 37-kDa FXIIIA, we analyzed its cellular localization under normal conditions and conditions where in vitro bone formation was inhibited. MC3T3-E1 osteoblasts were grown for 6 days in conditions where cells form collagen matrix and mineralize (AA 1 βGP treatment). Cells were subjected to subcellular fraction-ation that separated nuclear, cytosolic, membrane, and cytoskeletal proteins. To inhibit in vitro bone formation, cells were also additionally treated with a bone formation inhibitor, forskolin, which activates protein kinase A by increasing intracellular cyclic AMP levels. This inhibits Cbfa1 transcription factor, which is critical for osteoblast differentiation (Tintut et al. 1999; Li et al. 2004). As demonstrated in Figures 7A and 7B, forskolin treatment blocks mineralization and collagen deposition in the cultures. Analysis of FXIIIA expression in control osteoblast cultures showed that 37-kDa FXIIIA is found mainly in the cytosol and on the cell membrane. Treatment with forskolin resulted in a dramatic change in FXIIIA expression; upon this treatment, enzyme was found as the full-length 80-kDa form. Most interestingly, FXIIIA was found mainly in the nucleus and the cytosol, and only negligible amounts were present on the cell membrane. These data clearly show that full-length FXIIIA is expressed by the cells but is rapidly processed to its smaller form via a process regulated by protein kinase A. The different subcellular localization of full-length and 37-kDa FXIIIA indicates that they have different protein characteristics. Moreover, this strongly suggests that FXIIIA processing to the 37-kDa form is linked to promotion of osteoblast differentiation.
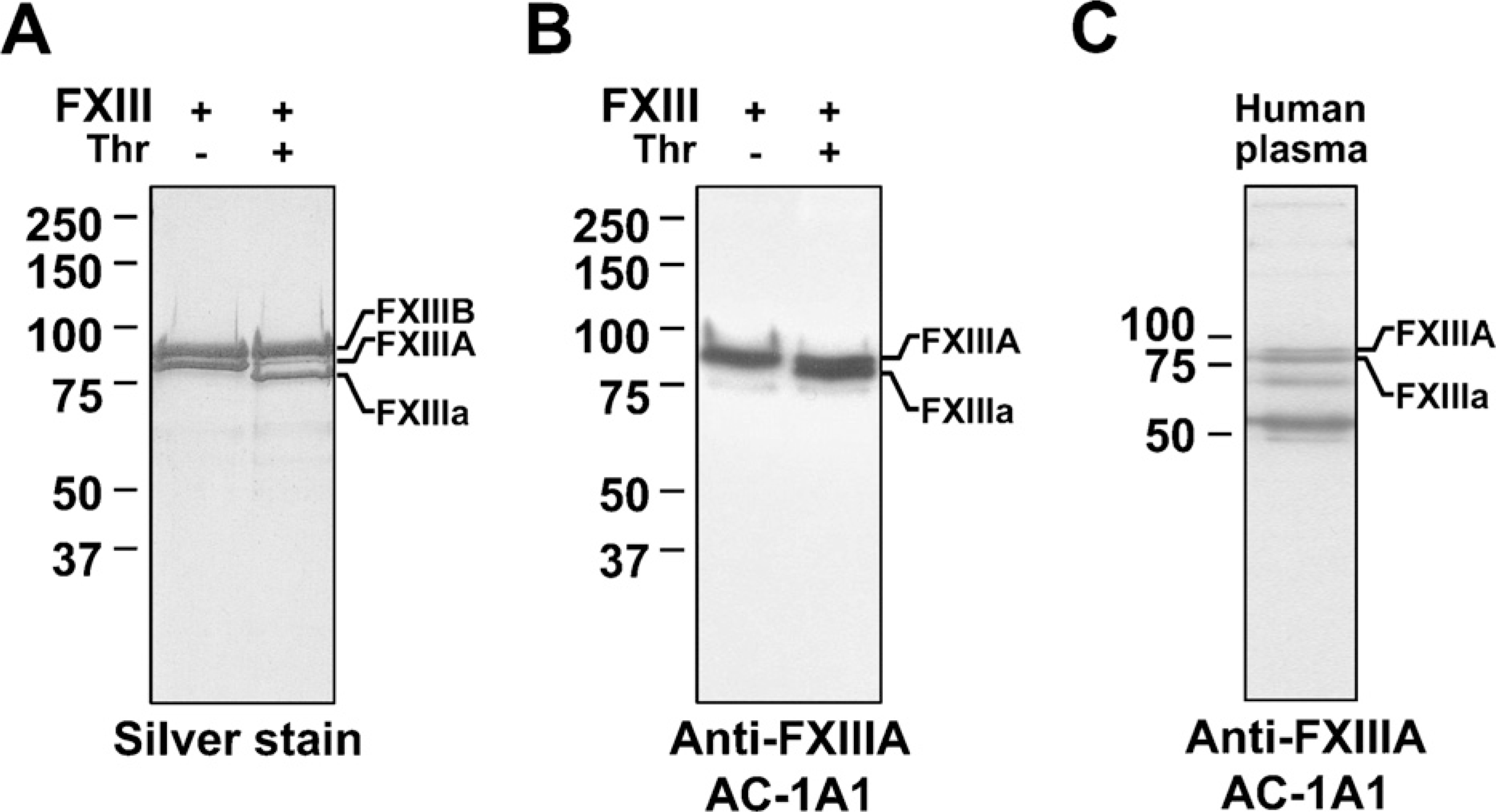
Specificity of monoclonal FXIIIA antibody (AC-1A1) in Western blotting. (
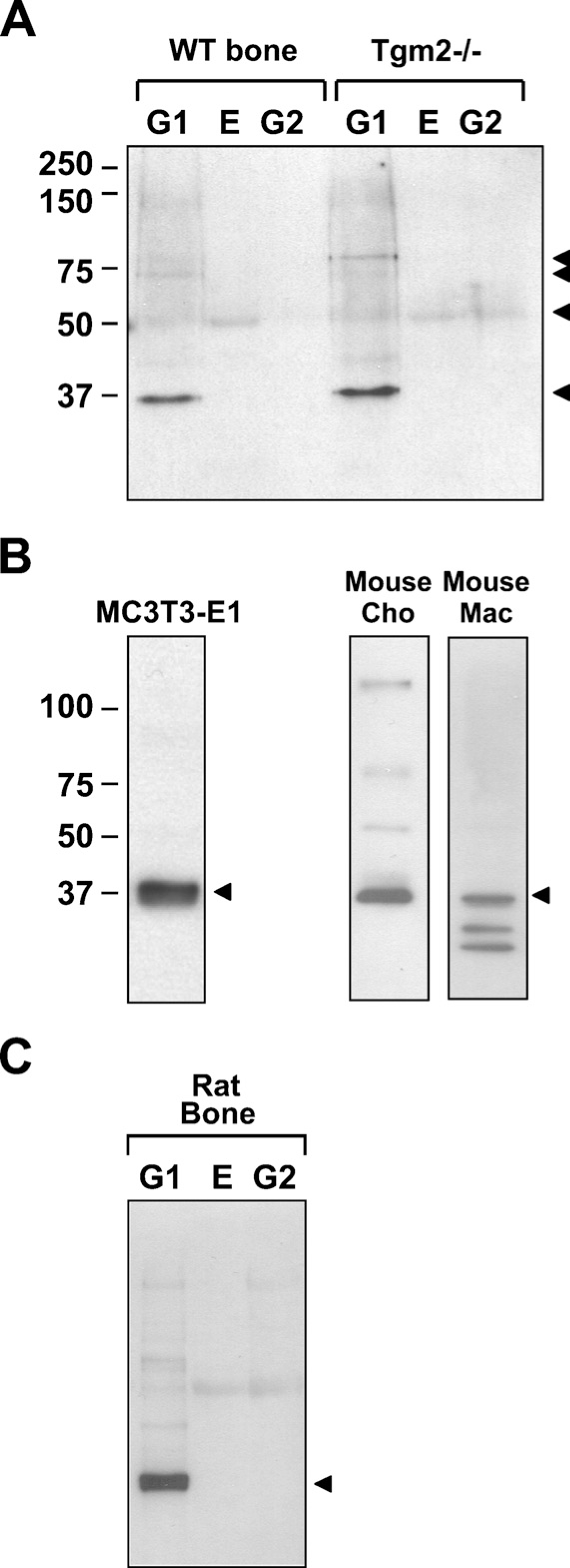
Western blot analysis of FXIIIA production in long bones (tibia and femur) of wild-type and Tgm2−/- bones and cellular extracts of osteoblasts, chondrocytes, and macrophages using monoclonal FXIIIA antibody AC-1A1. (
Discussion
In this study we show expression of plasma TG (FXIIIA) in mouse bones using IHC, in situ hybridization, and biochemical methods. FXIIIA mRNA was expressed in both osteoblasts and osteocytes, and the protein was found in both cell types and also in the osteoid layer of bone. Observation by light microscopy revealed the strongest FXIIIA mRNA message and most intense protein localization in osteoblasts and in young osteoid osteocytes in long bone. This expression pattern is similar to that seen for TG2, with the exception that TG2 appears to additionally accumulate in the pericellular matrix of osteocytes (Kaartinen et al. 2002). FXIIIA protein was found in bone cells and in bone matrix mainly as a 37-kDa form. This same form was also found in mouse chondrocyte and macrophage extracts but not in plasma, implying that 37-kDa FXIIIA could represent the tissue and cell form of FXIIIA. This form is also present in rat bone extracts, indicating that the form is not mouse specific. It remains unknown whether it is also found in human bone tissues or osteoblast cells. All cell and tissue extracts were prepared in protease inhibitor cocktails and kept cold at all times; thus, it is unlikely that the smaller form is as a result of protein degradation after extraction. Mouse chondrocyte extract was prepared by a different academic laboratory, and macrophage extract was obtained commercially, further lessening the possibility that the 37-kDa FXIIIA would result from degradation during purification procedures. We showed by RT-PCR analysis that the full FXIIIA gene is transcribed in osteoblast cells and no smaller mRNA variants are present, indicating that the 37-kDa form results from proteolytic prosessing. To the best of our knowledge, the presence of this lower molecular mass form of FXIIIA has not been reported for any other tissue or cell culture system. Reports showing FXIIIA in Western blots of articular or growth plate cartilage describe full-length FXIIIA (Recheis et al. 2000; D'Argenio et al. 2001; Nurminskaya et al. 2002; Undas et al. 2003; Rosenthal et al. 2004). Based on the fact that we were able to detect 37-kDa FXIIIA in chondrocyte and macrophage extracts, it is likely that the monoclonal antibody used here detects this form particularly well. Since the antibody clearly also detects full-length FXIIIA, it is not specific to the smaller form. Although the characteristics of the 37-kDa FXIIIA remain unknown, the fact that the AC-1A1 antibody epitope is in the calcium-binding region (amino acids 462-Lys to 501-Gly) of FXIIIA indicates this region must be present in the 37-kDa form. Also, because 37-kDa FXIIIA is not sensitive to thrombin digestion, thrombin cleavage sites are not likely present. The observations that bone and osteoblast extracts do contain thrombin-activated FXIIIA indicate that both full-length and 37-kDa FXIIIA coexist in bone and osteoblasts (Al-Jallad et al. 2006; Nurminskaya and Kaartinen 2006). This idea is also supported by our Western blot data on bone extracts that show small amounts of full-length FXIIIA. Whether the 37-kDa form is or can be activated by other proteases remains unknown. Studies are currently in progress to characterize the 37-kDa FXIIIA form in greater detail. Further evidence for the relevance of the 37-kDa FXIIIA in bone formation derives from our data showing that it is found in the cytoplasm and at the plasma membrane of osteoblasts undergoing differentiation (collagen deposition and mineralization). In the cell cultures where osteoblast differentiation and in vitro bone formation were inhibited by the protein kinase A activator forskolin, FXIIIA was expressed in its full-length form that was less associated with plasma membrane. This implies that FXIIIA processing to the 37-kDa form is related to the normal osteoblast differentiation process and that this processing likely allows the enzyme to localize and bind to the plasma membrane, thus the localization at the cell surface. The observation that the 37-kDa form is also located in macrophages and chondrocytes indicates that the form is not osteoblast specific. This points to a general mechanism of how FXIIIA could be involved in osteoblast differentiation. Indeed, cell surface FXIIIA found in several cell types such as platelets and macrophages has been linked to cytoskeletal organization, which is a critical cellular event and requirement for normal cell activity including cell differentiation (Adany and Bardos 2003).
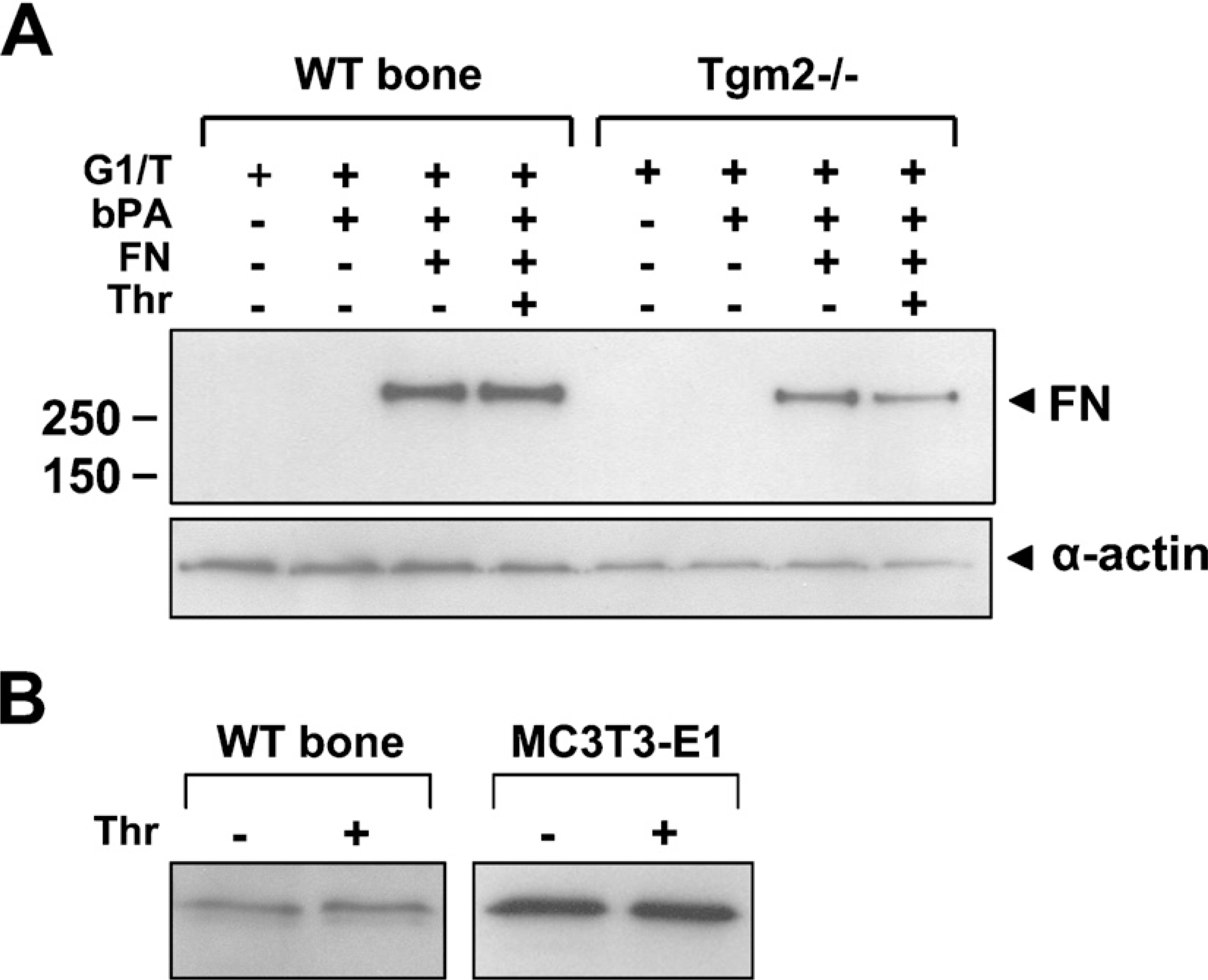
Transglutaminase (TG) activity in wild-type and Tgm2−/- mouse bones. (
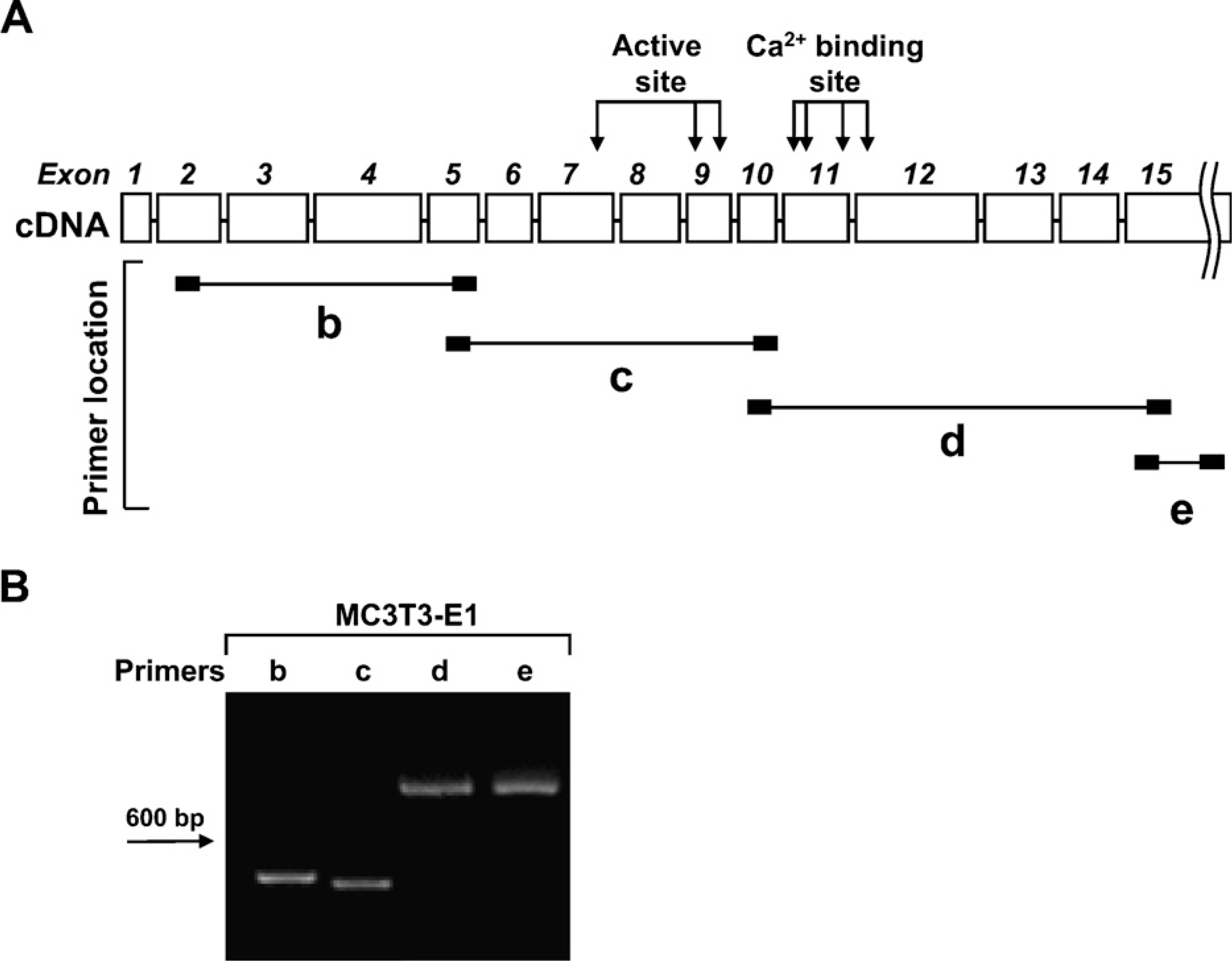
RT-PCR strategy to screen for FXIIIA splice variants in MC3T3-E1 osteoblasts. (
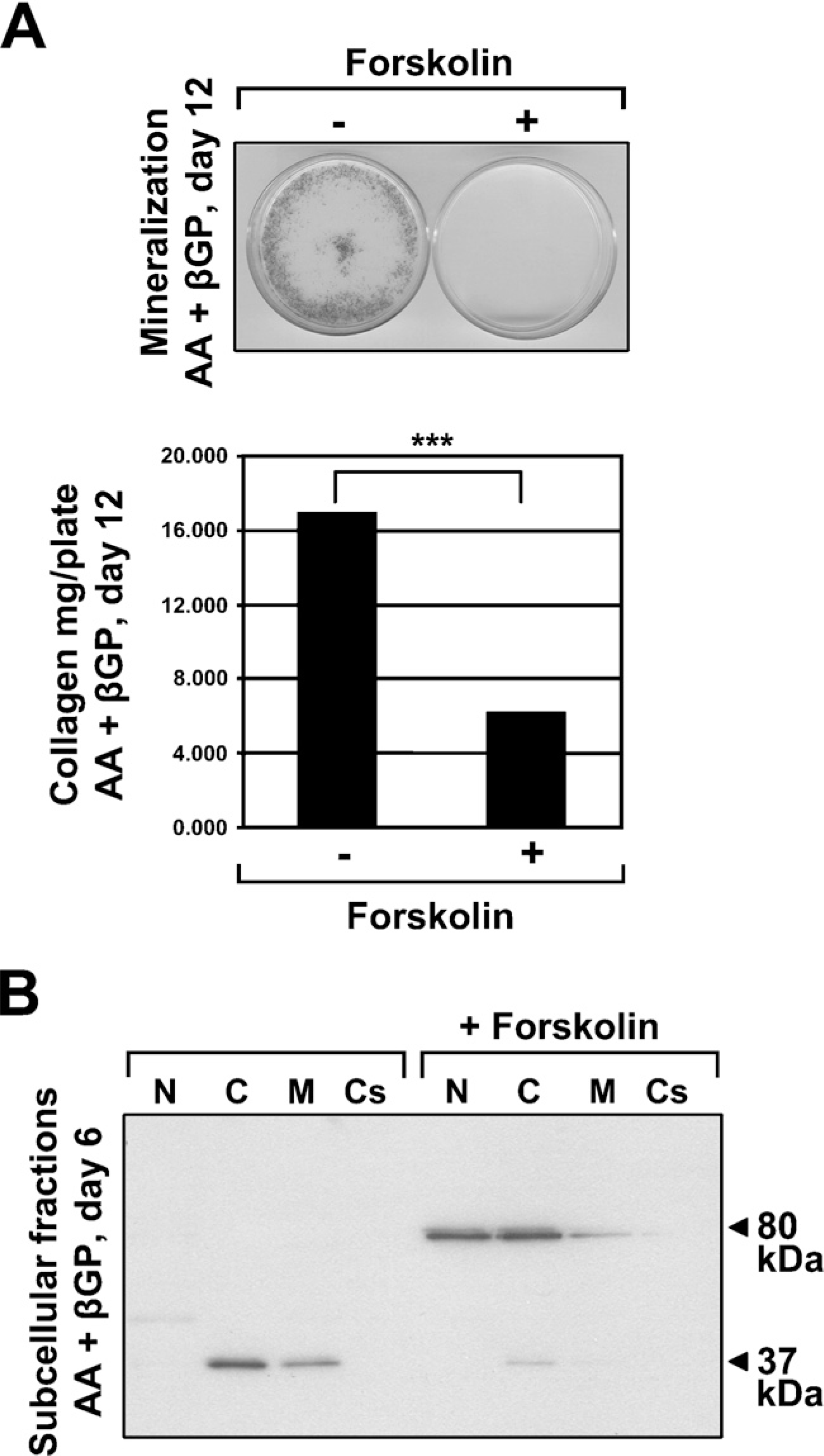
Protein kinase A-regulated processing of FXIIIA and association with the plasma membrane. (
The role of FXIIIA in connective tissues is still largely unknown; however, many investigators have shown that TG activity in general can regulate matrix production in vitro and in vivo, and the role of FXIIIA in this process is becoming increasingly evident. FXIIIA-null mice show significantly decreased COL I deposition during cardiac remodeling after myocardial infarction (Nahrendorf et al. 2006). Similarly, inhibition of TG activity in kidney tubular cells reduces glucose-induced ECM accumulation (Skill et al. 2004), and causes a dramatic decrease in COL I deposition in osteoblast cultures (Al-Jallad et al. 2006). Moreover, our cell culture studies suggest that FXIIIA has a more prominent role in matrix formation than TG2. During the osteoblast differentiation program where cells deposit a COL I-rich matrix that later mineralizes, TG2 levels remain similar throughout the culture period. FXIIIA levels, on the other hand, steadily increase and correlate with the expression of osteoblast differentiation markers during the differentiation process (Al-Jallad et al. 2006). TG2 is found on the cell surface and partly in the ECM where it remains throughout cell differentiation in the cultures, whereas FXIIIA expression responds (increases) to AA treatment, which in MC3T3-E1 cell cultures is a critical supplement for COL I deposition. Upon AA treatment, MC3T3-E1 cells externalize FXIIIA to the cell surface and to the ECM. These observations are in complete agreement with the work presented in this study showing that proteolytic cleavage of FXIIIA and association of its smaller 37-kDa processed form with the osteoblast membrane correlates with osteoblast differentiation stage. Because initial COL I deposition is a critical event for further progression of osteoblast differentiation program, it is possible that cell surface FXIIIA is an important player in cell-matrix communication during the initial steps where FN and COL I network is formed. It is also likely that cell surface FXIIIA is also secreted into the ECM. TG activity assays using conditioned medium from AA-treated cells showed that the main protein incorporating the primary amine label was exactly a protein having a molecular mass of tilde;33–37 kDa; indeed, likely the small FXIIIA form itself (Al-Jallad et al. 2006). The fact that FXIIIA immunostaining was weak or absent from bone tissue ECM, as shown in the present study, at first glance seems contradictory to the cell culture results demonstrating its secretion. However, because staining was observed (albeit weakly) for FXIIIA in the osteoid, it is possible that changes associated with mineralization of the matrix prevents its IHC detection in the ECM by masking antibody epitopes. Alternatively, the protein might be removed or completely degraded from the osteoid just prior to its mineralization. A similar staining pattern was shown for TG2 in bone where it was found in the ECM of the osteoid layer but not in the mineralized matrix (Kaartinen et al. 2002).
In summary, we present novel in vivo and in vitro data showing expression of FXIIIA by osteoblasts and osteocytes in bone and in bone cell cultures. FXIIIA localizes to the newly deposited osteoid layer of bone, consistent with a role in early matrix formation. FXIIIA is mainly produced by bone cells as a 37-kDa form, which results from posttranslational proteolytic processing that can be inhibited by the protein kinase A. Moreover, because the 37-kDa form was found only in cells and not in plasma, it is possible that it represents the tissue and cellular form of FXIIIA. In tissues and in bone cell culture, it appears that the 37-kDa form of FXIIIA associates with the osteoblast plasma membrane as part of the osteoblast differentiation process.
Footnotes
Acknowledgements
This study was supported by grants (to MTK) from the Canadian Institutes of Health Research (CIHR) and the CIHR Institute for Musculoskeletal Health and Arthritis and from the Fonds de la Recherche en santé du Québec. Training grants were received from the CIHR Training Program for Skeletal Health Research (to HFA-J) and from the CIHR Strategic Training Program in Applied Oral Health Research (to YN).
The authors thank Dr. Gerry Melino (University of Tor Vergata, Rome, Italy) for the Tgm2−/- knockout mice and Dr. Monzur Murshed (Baylor College of Medicine, Houston TX) and Dr. Peter Davis (MD Anderson Cancer Center, Houston, TX) for generously preparing the Tgm2−/- tissues for this work. We also thank Dr. René St-Arnaud (Shriners Hospital and Department of Human Genetics, Faculty of Medicine, McGill University, Montreal, QC, Canada) for donating primary osteoblast RNA, and Dr. Maria Nurminskaya (Tufts University, Department of Anatomy and Cell Biology, Boston, MA) for kindly donating the embryonic mouse chondrocyte extract.