
Abstract
In situ hybridization (ISH) is a particularly useful method to investigate de novo mRNA expression in tissue sections. High specificity and sensitivity of this technique combined with the great preservation of tissue and cellular morphology conferred by fixatives such as 4% paraformaldehyde, pH 9.5, make ISH a tool of choice for detecting genes of interest in individual cells in the central nervous system (CNS). Here we describe a novel method that combines radioactive ISH with immunofluorescence on the same tissue section to identify cell populations expressing selected mRNA transcripts. This novel method has several major advantages over previously described double-labeling light microscopic methods combining enzymatic immunohistochemistry and ISH including (1) complete protection against loss of hybridization signal that normally occurs during the immunoenzymatic reaction, (2) improved immunolabeling sensitivity due to the proteinase K digestion step during ISH, (3) detection of several proteins specific for different cell populations on the same tissue section, and (4) counterstaining of tissue sections without affecting visualization of immunolabeling. This new method will be particularly useful for investigators looking to identify cell populations producing mRNAs expressed in low abundance such as cytokines, chemokines, and growth factors in the intact and/or injured mammalian CNS.
Keywords
I
One way of investigating de novo gene expression in tissue sections is to use in situ hybridization (ISH). ISH offers many advantages over most other techniques used for detecting target mRNAs in tissue samples (e.g., RT-PCR, RNase protection assay, and Northern blot): (1) high specificity compared with RT-PCR, (2) visualization of the spatial distribution of target mRNAs at the single cell level, and (3) identification of cellular populations that express target mRNAs by combining, on the same tissue section, ISH with histochemistry, immunohistochemistry (IHC), neuronal tract tracing, or even ISH (e.g., combination of radioactive and nonradioactive probes).
Over the last two decades, many studies have combined ISH to IHC to identify cellular populations that synthesize a gene of interest. In these studies, cell-specific antigenic markers were first detected using immunoperoxidase reactions, followed by detection of target mRNAs using radioactive or non-radioactive probes. We have found, however, that peroxidase reaction products cause a decrease in mRNA signal, and this can seriously compromise detection of low copy number mRNA transcripts such as those coding for cytokines and chemokines. This situation is even more problematic in models in which cells enriched in endogenous peroxidase are recruited at the lesion/disease site(s). As previously demonstrated by us and others (Bohatschek et al. 2001; Lacroix et al. 2002), leukocytes rich in endogenous peroxidase can be recruited in large numbers in the CNS following insult and even remain in the area for prolonged periods of time, depending on the cell type and the model being studied.
Thus, the apparent incompatibility between ISH signal and enzymatic reaction products has prompted us to develop a new methodology that allows for the combination of ISH with immunofluorescence (IF) to identify CNS cell types expressing selected mRNA transcripts. We also present a series of histochemical data that clearly demonstrate why our novel approach is superior to the more traditional double-labeling technique combining IHC to ISH. Finally, we show examples of multiple labeling combining ISH for the detection of low-abundance cytokine and chemokine mRNAs with several IF labels on the same CNS tissue sections.
Materials and Methods
Animals
Overall, a total of 40 mice and rats were used in this study. Adult female C57BL/6 mice (8-12 weeks old) weighing 18-20 g and adult male Sprague Dawley rats (8-10 weeks old) weighing 200-225 g were obtained from Charles River Laboratories (Montréal, QC, Canada). Mice and rats had free access to food and water. All surgical procedures were approved by the Laval University Animal Care Committee and followed Canadian Council on Animal Care guidelines. The experimental protocol included three groups of animals: naive (n = 4), sham operated (n = 10), and SCI (n = 26).
Experimental SCI
C57BL/6 female mice were deeply anesthetized with isoflurane and underwent laminectomy at vertebral level T9-10, which corresponds to spinal segment T11-12. Briefly, the vertebral column was stabilized and a contusion of 70 kdyn was performed using the Infinite Horizon SCI device (Precision Systems and Instrumentation; Lexington, KY). Overlying muscular layers were then sutured and cutaneous layers stapled. Postoperatively, animals received manual bladder evacuation twice daily to prevent urinary tract infections. SCI mice were sacrificed by perfusion at 1, 3, 6, and 12 hr and 4 and 14 days postcontusion.
Tissue Preparation
Mice and rats were overdosed with a mixture of ketamine and xylazine and transcardially perfused with cold 0.9% saline solution followed by 4% paraformaldehyde (PFA), pH 9.5, in Borax buffer. After perfusion with the fixative, spinal cords were dissected out, postfixed for 2 days, and placed overnight in a 4% PFA-Borax/10% sucrose solution. Spinal cords were cut into 4-mm segments. For each animal, a total of three spinal cord segments were cut using a cryostat (Leica Microsystems; Richmond Hill, Ontario, Canada). The first segment was centered at the site of trauma and included 2 mm on each side of the lesion. Rostral and caudal 4-mm segments were located 2-6 mm distal to the injury site, on each side of the lesion. The day of the sectioning, spinal cord segments were placed side by side, rostral end facing up, into plastic molds and covered with Tissue-Tek embedding medium (OCT compound; Canemco, St-Laurent, Québec, Canada). The entire mold was then quickly frozen over dry ice and left in place until tissue processing. Thirty-μm-thick (mouse) and 14- and 30-μm-thick (rat) cryostat coronal sections were collected directly onto slides with a permanent positively charged surface (Surgipath Canada; Winnipeg, Manitoba, Canada) and separated into seven different series of adjacent sections. Slides were stored at −20C until ISH, IF, or multiple labeling was performed.
cRNA Riboprobes
ISH was carried out to detect the mRNAs coding for the following cytokines and chemokines: TNF-α, IL-1β, IL-6, LIF, MCP-1, MIP-1α, and fractalkine. Full-length cDNAs for mouse TNF, IL-1β, IL-6, LIF, and MCP-1 and rat fractalkine cloned into expression vectors pBluescript II SK+ (IL-6, TNF), pCRII-TOPO (IL-1β), pcDNA (LIF), pGEM-1 (MCP-1), and pGEM-7 (fractalkine) were obtained from Dr. Serge Rivest (Laval University; Québec, Canada). The full-length cDNA coding for mouse MIP-1α was kindly provided by Dr. Barbara Sherry (North Shore-Long Island Jewish Research Institute; Manhasset, NY). For MIP-1α, coding sequence corresponding to nucleotides 1-279 was amplified using the following primers: forward, 5î-gcatcgaattcatgaaggtctccaccactgcc-3î, reverse, 5î-ggtcactcgagtcaggcattcagttccaggtc-3î. Coding sequences chosen for probe synthesis were selected to match only the intended genes as verified by BLAST search in Genbank. PCR products were subcloned into pBlue-script II KS+ at the EcoRI-XhoI restriction sites and then sequenced to confirm gene identity. After linearization of the plasmids, radiolabeled cRNA probes were synthesized using the Riboprobe Combination System SP6/T7 and T3 RNA polymerase (Promega; Madison, WI) according to a protocol published by Simmons et al. (1989). More specifically, riboprobes were synthesized by incubation of 250 ng of the linearized plasmid in 6 mM MgCl2, 40 mM Tris (pH 8.0), 2 mM spermidine, 10 mM NaCl, 10 mM dithiothreitol (DTT), 0.2 mM ATP/GTP/CTP, 100 μCi [α-35S]UTP (NEN Life Science; Boston, MA), 40 U RNase inhibitor, and 20 U RNA polymerase for 60 min at 37C. Unincorporated nucleotides were removed using the ammonium-acetate method. DNase was added and phenol-chloroform extraction performed 10 min later. cRNA was precipitated using 5 M ammonium acetate (pH 8.0) and 100% ethanol for 20 min on dry ice. The pellet was washed with 70% ethanol, dried, and resuspended in Tris-EDTA. A concentration of 107 cpm probe was mixed into 1 ml of hybridization mixture (50% formamide, 10% dextran sulfate, 1X Denhardt's solution, 0.3 M NaCl, 10 mM Tris (pH 8.0), 1 mM EDTA (pH 8.0), 0.5 mg/ml tRNA, 10 mM DTT). This solution was mixed and heated for 5 min at 65C before being spotted on slides.
ISH
All sections were prehybridized, hybridized, and posthybridized in parallel to equalize background intensity. Slides were first removed from the freezer and thawed overnight at room temperature under vacuum. Next, tissue sections were fixed in 4% PFA for 20 min and digested for 25 min with proteinase K (PK; 10 μg/ml). Tissue sections were then rinsed in diethyl pyrocarbonate H2O, acetylated in 0.25% acetic anhydride in 0.1 M triethanolamine, pH 8.0, and dehydrated through graded concentrations of alcohol. After vacuum drying, 100 μl of hybridization mixture (107 cpm/ml) was spotted on each slide, sealed under a coverslip, and incubated for ~15-18 hr on a slide warmer preheated at 60C. Slides were rinsed in 4X standard saline citrate (SSC) at room temperature for 30 min and then coverslips were gently removed. After additional rinses in 4X SSC, tissue sections were digested with RNase A (20 μg/ml, 37C, 30 min), rinsed in decreasing concentrations of SSC (2X, 1X, 0.5X), and then washed in 0.1 × SSC for 30 min at 60C. Tissue sections used for double or multiple labeling were rinsed in potassium-PBS (KPBS) for 5 min and immediately processed for IHC as described below. Tissue sections used for ISH alone were rinsed in 0.1X SSC, dehydrated through graded concentrations of alcohol, defatted in Hemo-D (Fisher Scientific), and finally dipped in NTB nuclear emulsion (diluted 1:1 in distilled H2O; Kodak, Rochester, NY).
Combination of IHC With ISH
Spinal cord sections were processed for IHC staining using our previously published protocol, with the exception of the step in which tissue sections are incubated with 0.6% hydrogen peroxide (H2O2) in KPBS to reduce the activity of endogenous peroxidases (Lacroix et al. 1998,2002). The H2O2 quenching procedure was omitted in an attempt to target specifically the effects of immunoperoxidase reaction on mRNA signal loss. Briefly, tissue sections were successively incubated at room temperature with primary antibody for 2 hr, biotinylated secondary antibody (Vector Laboratories; Burlingame, CA) for 1 hr, and the avidin-biotinylated horse-radish peroxidase complex (ABC) solution (Vector Laboratories) for 1 hr. After rinsing in KPBS, sections were reacted for 2-10 min with the chromagen diaminobenzidine (DAB; Sigma, Mississauga, Ontario, Canada) to obtain a brown reaction product. Tissue sections were then dried for 1 hr under vacuum and processed for ISH as described above. During pre- and posthybridization and defatting steps, tissue sections were quickly dehydrated through graded concentrations of alcohol to avoid fading of the IHC staining. To determine if peroxidase reaction products can cause a loss of target mRNA signal already hybridized with riboprobes, one additional series of tissue sections was processed for ISH and then immunolabeled using the conventional ABC-immunoperoxidase approach.
Combination of ISH With Multiple IF Labeling
To identify the cell populations expressing target mRNA transcripts coding for a number of cytokines and chemokines, ISH was combined with IF. For this, tissue sections were first prehybridized, hybridized, and posthybridized as described above and then processed for multiple IF labeling. During the prehybridization step, PK digestion time was set to 25 min. IF labeling was performed using CoverWell incubation chambers (Invitrogen Canada; Burlington, Ontario, Canada) and the following protocol: (1) blocking for 1 hr in KPBS + 0.25% Triton X-100 + 5% normal goat serum, (2) incubation for 2 hr in primary antibodies at room temperature, (3) incubation for 2.5 hr in secondary antibodies conjugated to either the fluorophore Alexa-488 (1:200 dilution; Invitrogen Canada) or the fluorophore Rhodamine Red-X (1:200 dilution; Jackson ImmunoResearch, West Grove, PA), (4) counterstaining in DAPI (1:5000 dilution; Invitrogen Canada) for 20 min. The following antibodies were used to identify cell types: the ionized calcium-binding adaptor molecule 1 (Iba1) polyclonal antibody (1:750 dilution; Wako, Richmond, VA), the galactose-specific lectin-3 (galectin-3) monoclonal antibody (1:500 dilution; American Type Culture Collection, Manassas, VA), or the CD11b monoclonal antibody (1:200 dilution; Serotec, Raleigh, NC) for macrophages/microglia; the carbonic anhydrase II (CAII) polyclonal antibody (1:2000 dilution; a generous gift from Dr. Said Ghandour, Université Louis Pasteur, Strasbourg, France) for oligodendrocytes; glial fibrillary acid protein (GFAP) polyclonal antibody (1:1000 dilution; Dako, Mississauga, Ontario, Canada) for astrocytes; and the neuron-specific nuclear protein (NeuN) monoclonal antibody (1:250 dilution; Chemicon, Temecula, CA) for neurons.
After being rinsed successively in KPBS and H2O, sections were vacuum dried and then exposed at 4C to X-ray films (Amersham Biosciences; Baie d'Urfé, Québec, Canada) for 24 hr. The next day, sections were dipped in NTB nuclear emulsion diluted 1:1 in H2O at 42C (Mandel Scientific Company; Guelph, Ontario, Canada), dried for 2 hr in the dark, and exposed in the dark at 4C with desiccant for 14 days. Dehydration and defatting steps that normally precede dipping into nuclear emulsion were omitted to avoid unnecessary fading of the fluorescence. Two weeks later, slides were developed in D19 developer (Kodak) for 3.5 min at 14-15C, washed 15 sec in H2O, and fixed in rapid fixer (Kodak) for 5 min. Tissue sections were then rinsed in H2O, quickly dehydrated through graded concentrations of alcohol, rapidly cleared in hemo-D, and coverslipped with DPX (Cedarlane Laboratories; Hornby, Ontario, Canada). The presence of each mRNA transcript was detected by the agglomeration of silver grains within the cell cytoplasm. IF labelings were visualized with a fluorescent microscope using UV-excitation filter for DAPI and absorption spectra (μm) filters of band-passes 515-555 for Alexa-488 and 590 for Rhodamine Red-X. IF was then combined with darkfield illumination to simultaneously locate multiple cell populations with silver grains using autoradiography. Digital microscopic images were collected with a high-resolution Retiga QICAM fast color 1394 camera (1392 × 1040 pixels; QImaging, Burnaby, BC, Canada) installed on a Nikon Eclipse 80i microscope (Nikon; Mississauga, Ontario, Canada).
Quantification of ISH Signal and IHC Staining
MIP-1α mRNA expression, as detected by various labeling methods, was quantified by two different approaches: (1) measuring the average density of MIP-1α mRNA signal per cell, and (2) counting the number of cells expressing MIP-1α mRNA in coronal sections found within a specific distance interval from the lesion epicenter. Optical densities of ISH signal were measured at a magnification of X20 within a 13-μm circular frame positioned over individual hybridized cells, as determined by agglomeration of silver grains. Under brightfield illumination, the circular frame was first centered over the Nissl-stained nucleus of each positive cell randomly selected using stereology tools. The density of MIP-1α mRNA signal was then measured under darkfield illumination on video images of tissue sections transmitted by the Retiga camera (QImaging), using the Bioquant Nova Prime software (Bioquant Image Analysis; Nashville, TN). Optical densities were corrected for the average background signal, which was measured in three circular frames 13-μm in diameter and placed in regions located close to the positive cells. On average, optical density measurements were obtained from 28 positive cells per coronal section. This number corresponds to the total number of positive cells found in sampled areas, which under the settings used covered ~25% of the entire coronal sections (sampling frames of 200 × 200 μm equally spaced at 200-μm intervals). Two coronal sections located between 1260 and 1470 μm caudal to the lesion epicenter were analyzed. Data are presented as average density of mRNA signal per cell. For the second type of analysis, cells were visualized under darkfield illumination and then counted from evenly spaced coronal sections (n = 2) located between 1260 and 1470 μm caudal to the lesion epicenter at a magnification of X10. Cells were considered positive when the signal represented >4X the background value. Results were expressed as the average number of positive cells per cross-section. All quantifications were performed by a single observer. All data collection was done blind with respect to the identity of the treatments.
For quantification of Iba1 and CAII immunostaining and fractalkine mRNA signal, the proportional area of tissue occupied by labeling within a predefined region of interest (ROI) was measured as previously described (Lacroix et al. 2002). For each section analyzed (n = 3 sections per animal for Iba1 and CAII; n = 5 for fractalkine mRNA), a predefined sample box of 480 H × 640 W was first centered over the central canal of the spinal cord at X20 magnification. Video images of tissue sections transmitted by the Retiga camera (QImaging) were then thresholded using the Bioquant Nova Prime software such that only IHC-labeled product or ISH signal resulted in measurable pixels on the digitized image. Contrast between signal and background was maximized and held constant between all subjects. The area occupied by the signal within the ROI was then measured using the Bioquant Nova Prime software. Data were presented as the proportional area of the ROI occupied by Iba1 or CAII immunolabeling or fractalkine mRNA signal.
Results
This study was initiated to develop a new method that will allow the identification of cellular populations that synthesize low copy number mRNA species in fixed tissue sections. Development of such a method became necessary because of the finding that immunoperoxidase reaction, which is an essential step of the traditional double-labeling technique combining IHC to ISH, causes a decrease in mRNA signal, therefore rendering the colocalization of low-abundance mRNA transcripts within immunolabeled cells very difficult. To overcome this particular problem, we developed a novel multiple labeling method that does not involve immunoenzymatic reaction products.
Peroxidase Reaction Products Cause a Decrease in mRNA Signal
One of the most important findings of the present study is that immunoperoxidase reaction products cause a decrease in hybridizable signal. An example of the deleterious effects of peroxidase reaction products on mRNA signal is given in Figure 1. In this figure, ISH was used to detect MIP-1α mRNA signal, which we have found to be strongly expressed at 3 hr post-SCI (Pineau I and Lacroix S, unpublished data) and combined or not to Iba1 immunostaining to label macrophages/activated microglia. Immunolabeling for macrophage and activated microglia was chosen for this particular experiment because cells of myeloid origin are the most important source of MIP-1α at 3 hr post-SCI (Pineau I and Lacroix S, unpublished data). Five series of adjacent spinal cord sections taken from the same SCI mice (n = 3) were hybridized with MIP-1α riboprobes and ISH signal measured and compared for the following conditions: (1) ISH alone, (2) immunoperoxidase labeling followed by ISH, (3) ISH followed by immunoperoxidase labeling, (4) immunoperoxidase labeling performed without DAB followed by ISH, and (5) ISH followed by multiple IF labeling (Figure 1). Quantitative analyses revealed that MIP-1α mRNA signal was significantly weaker in sections processed using any of the double-labeling methods combining immunoperoxidase labeling with ISH compared with the other conditions tested. More specifically, the average density of MIP-1α mRNA signal per cell and the mean number of MIP-1α+ cells per section were decreased by 28% and 32%, respectively, in sections subjected to immunoperoxidase labeling followed by ISH when compared with sections subjected to ISH alone (Figure 1). Another important finding of this particular experiment is that immunoperoxidase reaction products also cause the loss of target mRNA signal already hybridized with riboprobes. This was somehow surprising considering the fact that cRNA-mRNA hybrids are considered to be very stable molecules. This particular finding emphasizes even more the destructive effects of peroxidase reaction products on mRNA signal integrity. The average density of MIP-1α mRNA signal per cell and the mean number of MIP-1α+ cells per section were reduced by 24% and 37%, respectively, in sections subjected to ISH followed by immunoperoxidase labeling compared with sections subjected to ISH alone. By comparing mRNA signal obtained in sections subjected to immunoperoxidase labeling followed by ISH with the signal obtained in sections subjected to ISH followed by immunoperoxidase labeling (no difference was seen between these groups), we were also able to conclude that RNases that act specifically on single-stranded RNA contribute minimally to the loss of mRNA signal occurring during the course of the traditional double-labeling method. However, although unlikely, these results do not preclude the potential presence of contaminating RNases that could cleave double-stranded RNAs.
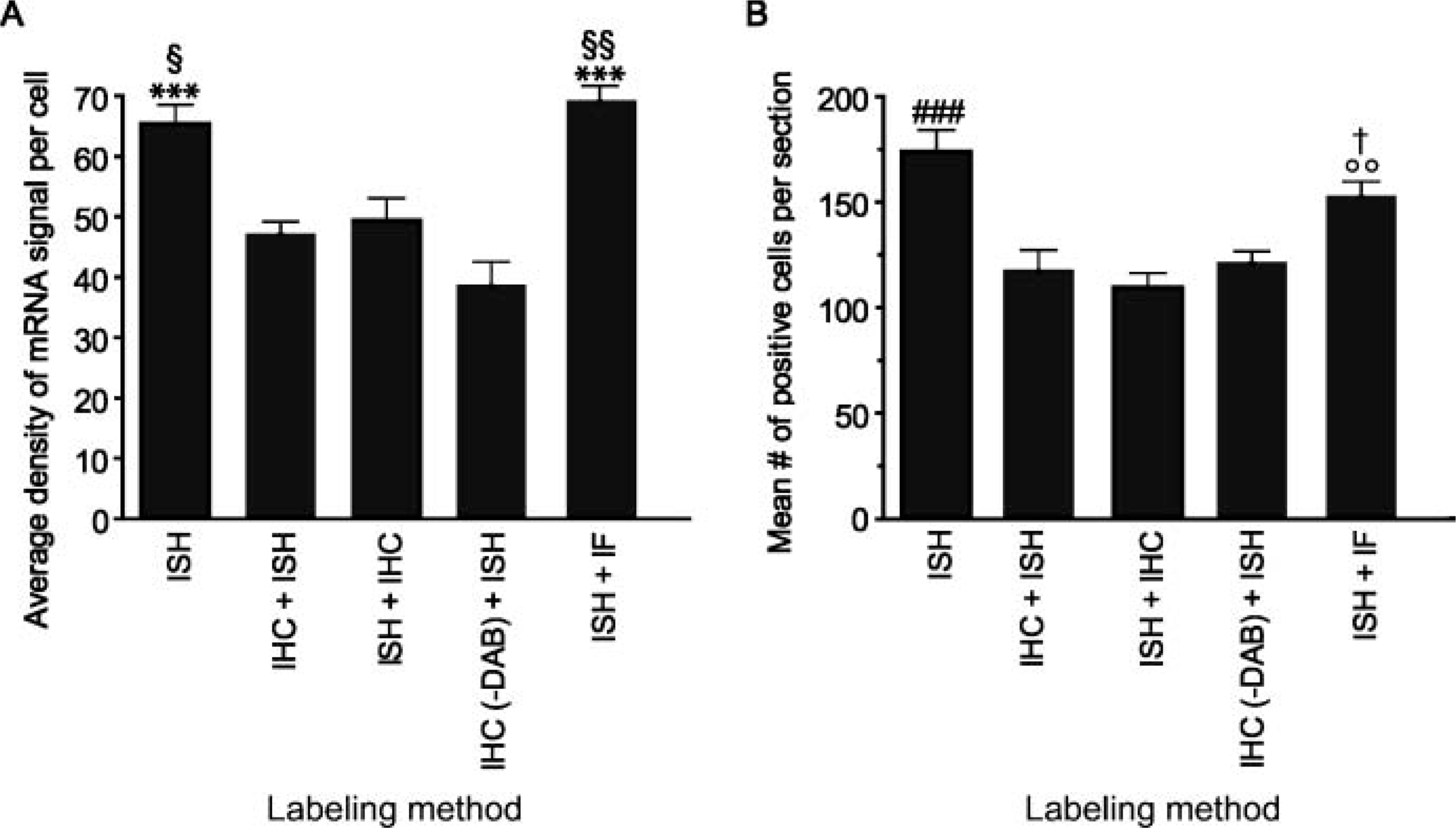
Peroxidase reaction products cause a decrease in mRNA signal. (
We next investigated the mechanism by which enzymatic reaction products could cause the disappearance of mRNA signal. Others have suggested that the chromogens used during the enzymatic reaction (e.g., DAB, NBT/BCIP, etc.) could form molecular aggregates with probes by intercalating in between bases (Chiu et al. 1996). To examine this possibility, one series of adjacent spinal cord sections was processed for immunoperoxidase labeling followed by ISH, except that DAB was omitted. Quantitative analyses revealed no significant difference in the average density of MIP-1α mRNA signal per cell and the mean number of MIP-1α+ cells for sections processed using any of the three protocols combining immunoperoxidase labeling with ISH. These results suggest that DAB contributes minimally to the loss of mRNA signal occurring during IHC. They rather indicate that free radicals generated during the immunoenzymatic reaction are responsible for the loss of mRNA signal. Together these findings highlight the importance of developing a novel multiple labeling method that does not involve immunoenzymatic reaction products, such as by combining IF with ISH or vice versa.
Pre-treatment of Tissue Sections With PK Improves IHC Staining
Compared with other methods of fixation, fixation of tissues by perfusion with 4% PFA buffered in Borax (pH 9.5) yields the strongest ISH signal using radioactive probes (Lacroix S, unpublished data). However, fixation in 4% PFA-Borax, pH 9.5, causes extensive cross-bridging between aldehydes and proteins, therefore reducing antibody penetration and antigen accessibility, which is already a problem when tissue sections thaw mounted onto slides are being used. As a result, immunodetection of antigens is often compromised by fixation with 4% PFA-Borax, pH 9.5. For example, we found that antibodies such as anti-Iba1, anti-CAII, anti-CD11b, and anti-NeuN, which all give very intense immunostaining on tissue sections fixed with 4% PFA-phosphate, pH 7.4, generate either weak immunostaining or no staining at all when used on tissue sections fixed with 4% PFA-Borax, pH 9.5. We therefore tested whether pretreatment of tissue sections with PK, a critical step of our ISH protocol, would facilitate the penetration and accessibility of the above-mentioned antibodies to their respective antigenic sites. Data are presented for one membrane antigen, the microglia/macrophage-specific marker Iba1 (Figures 2A-2D), and for one membrane/cytosolic antigen, the oligodendrocyte-specific marker CAII (Figures 2E-2H). Using 30-μ-thick adjacent spinal cord cross-sections taken from the same naive mice (n = 3), we compared immunostaining intensity after each of the following pretreatment conditions: (1) pretreatment with 0.25% Triton X-100, (2) pretreatment with Triton X-100 and incubation with PK at 10 μg/ml for 10 min, and (3) pretreatment with Triton X-100 and incubation with PK for 25 min. All sections were immunostained in parallel to equalize background intensity. As shown in Figure 2, pretreatment with PK allows the detection of antigens that were barely detectable (e.g., Iba1) or that could not be detected (e.g., CAII) with Triton X-100 pretreatment alone. Quantitative analyses revealed that, following incubation of tissue sections in PK for 10 min, signal intensity for Iba1 and CAII immunolabelings were increased by 18- and 134-fold, respectively, compared with pretreatment with detergent alone. It should be noted, however, that overdigestion with PK sometimes resulted in signal reduction and/or an increase in nonspecific background. Notably, we found that signal intensity for Iba1 immunolabeling was reduced by almost 3-fold following incubation of tissue sections in PK for 25 min compared with the 10-min treatment (Figure 2A). Although the intensity of Iba1 staining was significantly reduced following incubation of tissue sections in PK for 25 min, long and ramified processes of microglia were nevertheless easier to observe after 25 min of treatment (see insets in Figures 2B-2D). Time of enzymatic digestion must therefore be optimized for each antigen of interest. As a general rule, for 30-μm-thick tissue sections our results indicate that the optimal digestion time is 10 min to detect cell surface and membrane antigens and 25 min for cytosolic and nuclear antigens. For 14-μm sections, our observations suggest to limit proteolytic digestion to 5 min for cell surface and membrane antigens and to 10 min for cytosolic and nuclear antigens.
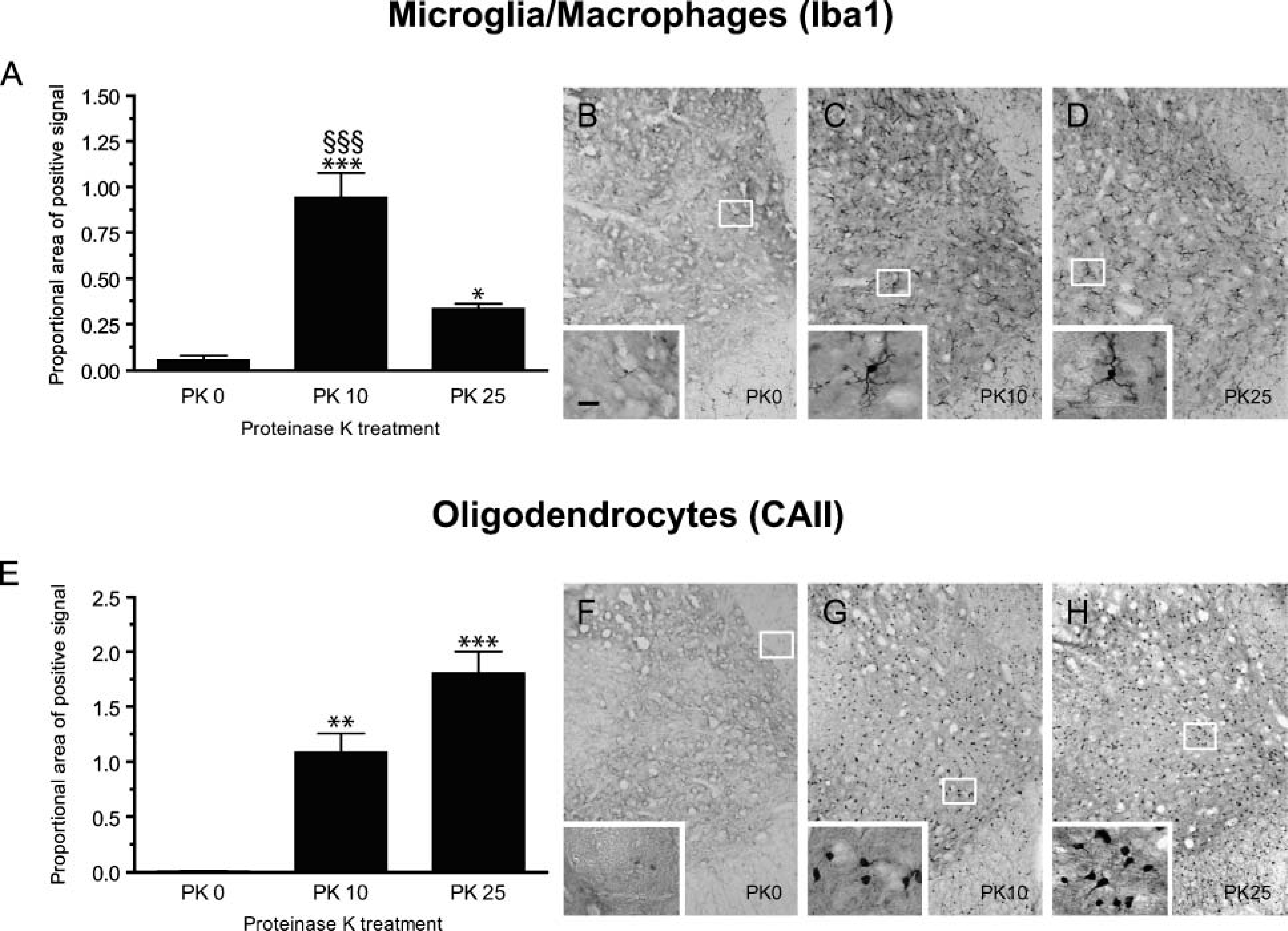
Proteinase-K (PK) pretreatment improves immunolabeling efficiency in central nervous system (CNS) mouse tissue sections fixed with 4% paraformaldehyde (PFA)-Borax, pH 9.5. (
Because pretreatment of tissue sections is believed to be important to expose target mRNAs to probes, we next investigated how changes in PK digestion time would affect ISH signal. For this particular set of experiments, we chose to examine mRNA expression of fractalkine, a chemokine reported to be constitutively expressed by most spinal cord neurons in naive rats (Verge et al. 2004). As demonstrated in Figure 3, proteolytic digestion of tissue sections is absolutely required to obtain maximal ISH signal. Results show that for 30-μm-thick tissue sections, omitting pretreatment or pretreatment of tissue sections in 0.25% Triton X-100 for 20 min yields very weak and inconsistent ISH signal (Figures 3A, 3C, and 3D). In contrast, fractalkine mRNA signal increased gradually with PK digestion time to reach its maximum after 25 min of pretreatment (Figures 3A, 3E-3H). After 25 min of PK digestion, fractalkine mRNA signal was more than 80 times greater than in non-treated 30-μm-thick tissue sections. For 14-μm sections, fractalkine mRNA signal increased with time up to 10 min of PK digestion (Figure 3B). The proportional area occupied by fractalkine mRNA signal was almost 2-fold greater within spinal cord sections treated with PK for 10 min compared with either non-treated or detergent-treated tissue sections. However, ISH signal started to decrease after 15 min of PK digestion and even dropped to ~60% of control levels (i.e., non-treated sections) after 25 min of digestion. This suggests that excessive PK pretreatment may result in the loss of target mRNAs. Together these results indicate that digesting cellular proteins using PK, for example, increases mRNA signal significantly compared with other methods of pretreatment tested in this study. Based on our data, we recommend the following pretreatment conditions for double-labeling studies combining ISH with IHC in CNS tissue sections fixed with 4% PFA-Borax, pH 9.5: (1) digestion for 25 min in 10 μg/ml PK for 30-μm-thick tissue sections, and (2) digestion for 10 min in 10 μg/ml PK for 14-μm-thick tissue sections.
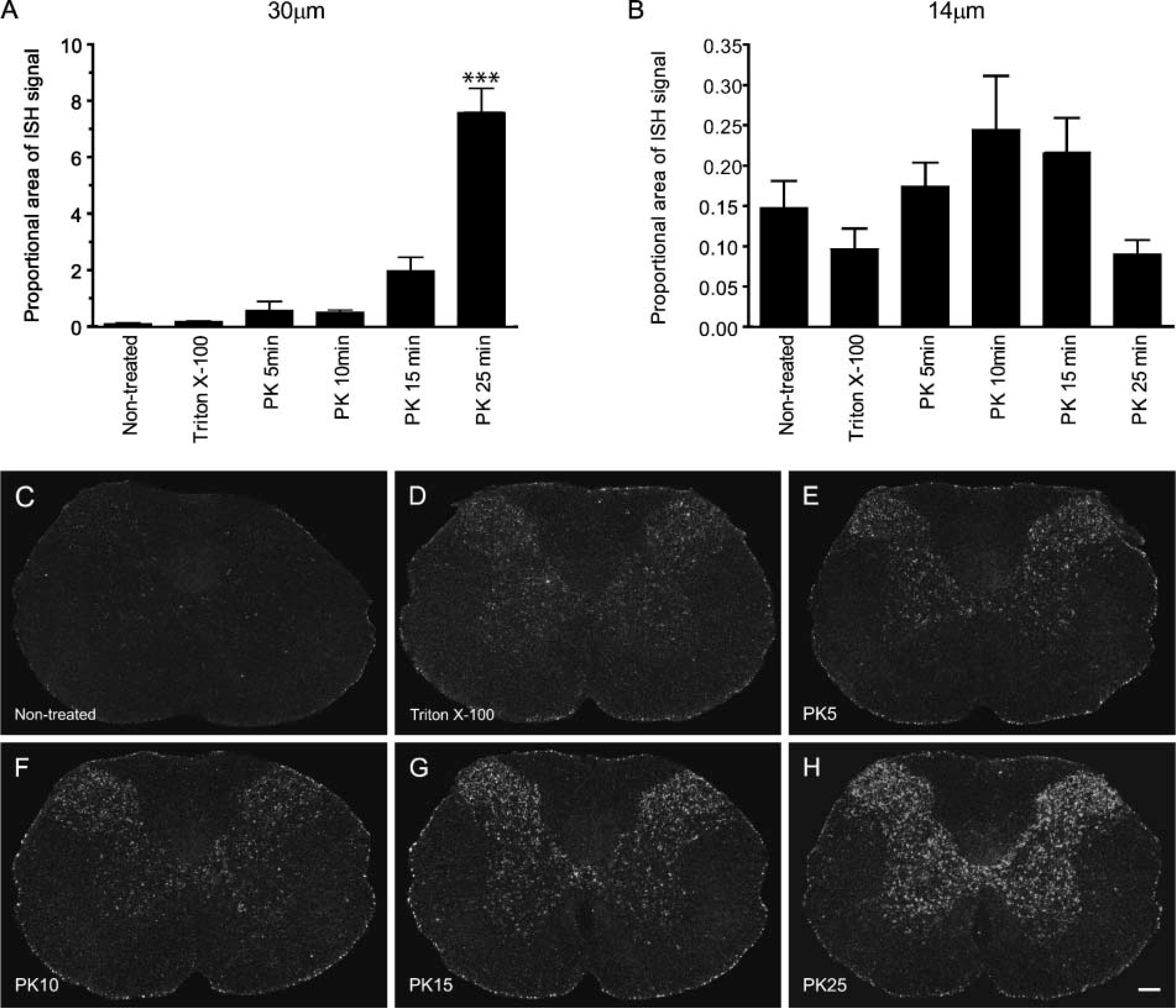
Pretreatment of PFA-fixed CNS tissue sections with PK is required to achieve maximal ISH signal. (
ISH Combined With IF Significantly Preserves mRNA Signal Compared With the Double-labeling Method Combining Immunoperoxidase Labeling With ISH
Quantitative analyses revealed that the ISH signal measured in sections subjected to ISH followed by multiple IF labeling was not statistically different from the signal observed following ISH alone (Figure 1). This suggests that IF labeling does not compromise the detection of target mRNAs. When compared with the traditional double-labeling method, the MIP-1α mRNA signal obtained using our new method was significantly higher. Specifically, the average density of MIP-1α mRNA signal per cell and the mean number of MIP-1α+ cells measured in sections processed using our new method corresponded to 146% and 130%, respectively, of the values detected in sections subjected to traditional double-labeling method involving immunoperoxidase labeling followed by ISH (Figure 1). These results highlight the advantage of our new procedure over the standard method with regard to the preservation of mRNA signal integrity.
Another significant advantage of combining ISH with IF is the possibility to simultaneously detect several antigens, corresponding to as many cellular populations on the same tissue section. Figure 4 shows examples of colocalization between low-abundance cytokine and chemokine mRNA transcripts such as LIF, MCP-1, IL-1β, TNF-α, IL-6, and various types of neural cells including GFAP-immunoreactive (ir) astrocytes (Figure 4A) and Gal-3-ir activated microglia/macrophages (Figures 4C and 4D). As demonstrated in this figure, an additional advantage of our method is that tissue sections can be counterstained with fluorescent reagents that specifically bind nucleic acid such as DAPI. Note that all double-labeled cells are associated with a single nucleus, therefore excluding the possibility that other cells in the neighborhood might have been responsible for the expression of the transcript. Without performing nuclear counterstaining, we found that it was basically impossible to determine with certainty that a specific target mRNA was colocalized within a specific cell type. This was due primarily to the presence of multiple cellular layers in tissue sections and the accumulation ofcell clusters in the injured CNS. A clear example of this is given in Figures 4E and 4F, which show that GFAP-ir astrocytes appear to express IL-6 mRNA. However, when DAPI counterstaining is overlaid on the figure, we clearly see that the IL-6 mRNA signal belongs to another cell not colocalized with GFAP immunoreactivity. Together these results underscore the importance of using nucleic acid stains to visualize nuclei in colocalization studies performed on tissue sections.
Discussion
In this study we presented evidence that peroxidase reaction products cause a decrease in mRNA signal. We have also shown that loss of hybridization signal caused by the presence of immunoperoxidase reaction products seriously compromised double-labeling studies combining IHC and ISH, especially when detection of low-abundance target mRNAs is involved. To resolve this problem, we have developed a new method that allows for the combination of techniques such as ISH and IF without causing alteration of mRNA signal integrity. With the new method described in this report, we successfully identified the cells that synthesize specific cytokines and chemokines expressed at relatively low levels in the injured mouse spinal cord.
The necessity of protecting target mRNAs from enzymatic degradation that may occur during IHC labeling was first suggested two decades ago (Shivers et al. 1986). The main recommendation resulting from this particular study was to add RNase inhibitors to the primary and secondary antisera. This was based on the concept that serum contains RNase activity that may cause mRNA degradation. However, as demonstrated in the present study, peroxidase reaction products, and not RNase activity, appear to be mainly responsible for the loss of mRNA signal occurring during IHC steps. Another important finding of this study is that peroxidase reaction products also cause the loss of target mRNA signal even when protected by a full-length cRNA probe, as shown in double-labeling studies in which ISH was followed by IHC. Interestingly, other enzymatic reaction products such as alkaline phosphatase reaction products were also found to contribute to substantial loss of ISH signal (Young 1989; Ozden et al. 1990; Young and Hsu 1991; Chiu et al. 1996). Although the exact mechanism by which enzymatic reaction products cause the disappearance of mRNA signal remains obscure, some studies have suggested that some of the reagents involved during the enzymatic reaction (e.g., NBT/BCIP, DAB, etc.) could form molecular aggregates with probes by intercalating in between bases (Chiu et al. 1996). However, our data suggest that peroxidase activity rather than intercalating chromogenic substrates could be responsible for mRNA degradation. We therefore speculate that reactive oxygen species, present or generated during the immunoperoxidase reaction, such as H2O2 and hydroxyl radicals (-OH) could be responsible for the loss of mRNA signal observed using the traditional double-labeling method.
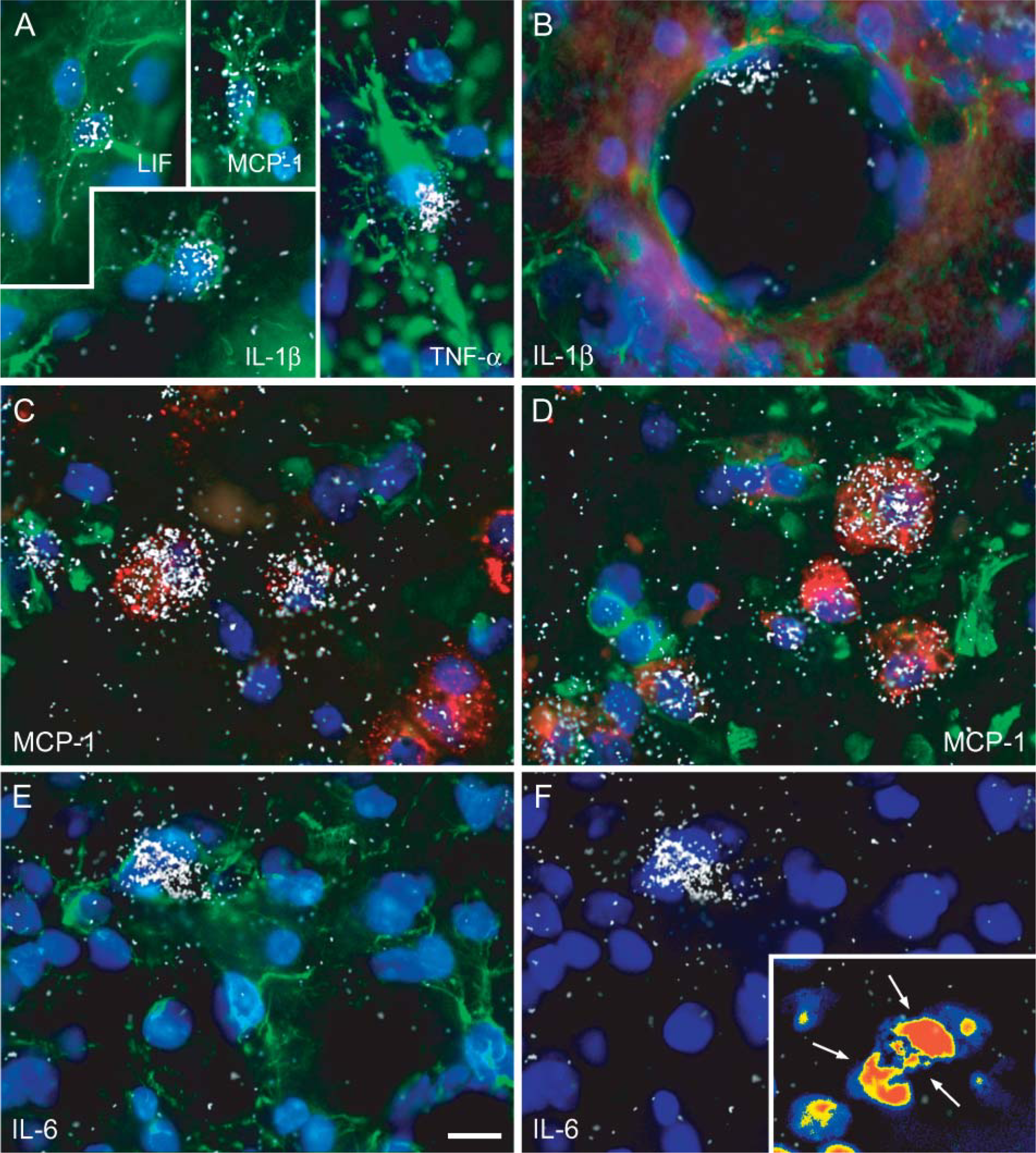
Examples of multiple labeling combining ISH with IF in the SCI mouse. (
It is important to point out, however, that the effects of immunoperoxidase activity on mRNA signal loss may not completely prevent colocalization of highly abundant mRNA species (e.g., mRNA transcripts coding for neuropeptides, enzymes, and receptors), as demonstrated by the abundant literature presenting colocalization data. In contrast, however, it can seriously compromise colocalization of rare mRNAs. A striking example of this is the fact that we were barely able to colocalize any cytokine and chemokine mRNAs within neural cells of the injured mouse spinal cord using the traditional method combining immunoperoxidase with ISH. The combination of ISH with IF did allow us to do that, as demonstrated by the detection of astrocytes and macrophages/microglia expressing cytokines/chemokines such as LIF, MCP-1, IL-1β, and TNF-α. It should also be mentioned that a decrease or loss of mRNA signal during IHC processing could have serious consequences for the interpretation of studies in which quantifications of double-labeled cells are reported. Among the consequences of the potential loss of hybridizable mRNA within peroxidase-immunolabeled cells are the absence of colocalization of a gene within a specific cell type(s) or an underestimation of the percentage and total number of double-labeled cells.
Although some studies have suggested that pretreatment of tissue sections may not always be necessary before ISH (Wahle and Beckh 1992), in our work optimized PK pretreatment always yielded a stronger ISH signal. In this regard, one specific study has suggested that PK digestion is particularly essential when lipid-rich tissues such as CNS and peripheral nervous system tissues are used (Schaeren-Wiemers and Gerfin-Moser 1993). Remarkably, our findings have also shown that optimized PK digestion of tissue sections fixed with 4% PFA-Borax, pH 9.5, confers a more intense immunostaining. Together these results highlight the importance of permeabilizing tissue sections before performing double-labeling studies combining ISH with IHC. Pretreatment with PK may also be useful to unmask antigenic sites prior to performing IHC on formaldehyde-fixed tissue sections. However, it is important to point out that PK digestion time must be optimized according to the thickness of tissue sections and the subcellular location of the antigen that we intend to reveal (e.g., outer membrane, cytosol, nucleus, etc.). It should also be mentioned that many other types of pretreatments have been documented for retrieving antigenicity or to allow for greater penetration of ISH probes including heating tissue sections (e.g., in microwave or autoclave), treatment with denaturing agents or acids, and digestion with enzymes other than PK (Shi et al. 1991,1993; Umemura et al. 1995; Oliver et al. 1997; Rait et al. 2004a,b; Yamashita and Okada 2005).
By combining the novel method described here and confocal microscopy, we have been able to detect ISH signal plus as many as four different fluorescent labels, corresponding to as many different cellular markers, simultaneously on the same tissue section. This is a major progress over double-labeling approaches because it enables the extraction of substantially more information from each tissue section. Maximizing the results obtained from each tissue sample will be particularly interesting for investigators working with animal models of CNS injury and diseases, especially considering that most of their research involves long-term studies and labor-intensive techniques, which translate into considerable costs. In our work, the ability to perform multiple labeling combining ISH with IF was particularly helpful to determine with certainty which glial cell type expresses cytokine and chemokine mRNA species. Another major advantage of our method is that it allows for nucleic acid counterstaining. In most other published protocols, counterstaining was not used because the coloration can sometimes obscure the visualization of immunolabeling and/or ISH signal. As demonstrated in Figure 4, counterstaining of tissue sections with a nuclear dye is absolutely essential for colocalization studies. Because of the presence of multiple cell layers in tissue sections, we found that it was very difficult to colocalize, with a certain degree of confidence, a target mRNA within a single cell without performing counterstaining. Of course, this exercise becomes almost impossible in injured and/or diseased CNS tissue sections, mostly because cells often coalesce in the injured/diseased CNS.
A rare limitation of our method over the traditional double-labeling method is the fact that long-term exposure of fluorophores to β-particle emissions of 35S accelerates fluorescence photobleaching. This can be avoided by the combination of non-radioactive ISH to IF. Compared with radioactive ISH methods, non-radioactive methods have some advantages such as safe use in the laboratory, longer shelf life of labeled probes, shorter signal development time, high resolution of signal, and longer time of storage of results. The fact that non-radioactive ISH methods are less sensitive and less specific than ISH using radiolabeled probes may explain why the radioactive procedure is still the method of choice. It should be pointed out, however, that methods have recently been developed to amplify non-radioactive ISH signals such as the tyramide/catalyzed reporter deposition amplification system (for reviews, see Speel et al. 1999,2006). The other possible disadvantages of our method include (1) the persistence of fluorescence signal—IF signal is not permanent and can bleach following excitation, and (2) the use of different microscope settings for darkfield/brightfield and fluorescence microscopy—the difficulty of visualizing both signals simultaneously can make quantification difficult. We have overcome these limitations by using fluorophores that are highly resistant to photobleaching (e.g., Alexa fluor dyes introduced by Molecular Probes) and by performing quantification on video images that are first captured and then overlaid using image analysis software.
In conclusion, our results highlight the importance of performing ISH before IHC during colocalization studies to preserve mRNAs. Pretreatment steps taking place during ISH do not compromise but rather improve IHC staining, although conditions must be optimized for each antigen of interest. Finally, because of the finding that peroxidase reaction products cause mRNA signal loss, we developed a novel method in which immunoperoxidase reaction was replaced by IF. This new technique yielded stronger ISH signal when compared with previous double-labeling methods and allowed for simultaneous detection of multiple antigenic markers on the same tissue section.
Footnotes
Acknowledgements
This study was supported by the Canadian Institutes of Health Research (CIHR). S.L. is supported by a Career Award from the Rx&D Health Research Foundation and CIHR.
We thank Nadia Fortin and Marthe Dubé for technical assistance. We are also grateful to Marc-André Laniel for help with editing this manuscript.