
Abstract
Cytokine-induced apoptosis inhibitor 1 (CIAPIN1) is a newly identified anti-apoptotic molecule. Our previous studies have demonstrated that CIAPIN1 is ubiquitously expressed in normal fetal and adult human tissues and confers multidrug resistance in gastric cancer cells, possibly by upregulating the expression of multidrug resistance gene 1 and multidrug resistance-related protein 1. However, fundamental biological functions of CIAPIN1 have not been elucidated. In this study, we first predicted the subcellular localization of CIAPIN1 with bioinformatic approaches and then characterized the intracellular localization of CIAPIN1 in both human and mouse cells by a combination of techniques including (a) immunohistochemistry and immunofluorescence, (b) His-tagged CIAPIN1 expression, and (c) sub-cellular fractionation and analysis of CIAPIN1 in the fractions by Western blotting. All methods produced consistent results; CIAPIN1 was localized in both the cytoplasm and the nucleus and was accumulated in the nucleolus. Bioinformatic prediction disclosed a putative nuclear localization signal and a putative nuclear export signal within both human and mouse CIAPIN1. These findings suggest that CIAPIN1 may undergo a cytoplasm-nucleus-nucleolus translocation.
Keywords
C
Proteins are evolved to function optimally in a specific subcellular localization, and the correct transportation of a protein to its final destination is crucial to its function. Therefore, revealing the subcellular localization of a specific protein often provides important information for the elucidation of its function. In the study conducted by Shibayama et al. (2004), immunofluorescence microscopy of mouse cells demonstrated that CIAPIN1 was localized exclusively in the cytoplasm. In our study on the survey of the distribution of CIAPIN1 in normal fetal and adult human tissues by immunohistochemical (IHC) staining with an in-house-produced anti-CIAPIN1 monoclonal antibody (MAb), we observed that CIAPIN1 was localized in the nucleus as well as in the cytoplasm (Hao et al. 2006b). This finding prompted us to investigate the subcellular localization of CIAPIN1 in both human and mouse cells. In the present study, subcellular localization of CIAPIN1 was verified using multiple techniques.
Materials and Methods
Tissue Specimens
Use of human tissues in this study was approved by the Institutional Review Board of the Fourth Military Medical University, Shaanxi Province, China and was done in accordance with international guidelines for the use of human tissues. Normal fetal tissues including skeletal muscle and colon and adult tissues including brain and gastric mucosa were from tissue microarrays EC01-001 and NC01-001 (Cybrdi; Xi'an, China), respectively. Fetal tissues were obtained from a normal 4- to 5-gestational day fetus. Adult tissues were obtained from a healthy 45-year-old man. All tissues were formalin fixed and paraffin embedded.
Antibodies
Mouse anti-CIAPIN1 MAb (clone 3C6) was developed by our laboratory (Hao et al. 2005). This antibody recognizes hCIAPIN1 and cross-reacts with mCIAPIN1. Anti-CIAPIN1 MAb used in this study was purified by protein G affinity chromatography. Mouse anti-6XHis tag (Ab18184, subtype IgG2b), GAPDH ((Ab9484), histone H1 (Ab14028), and fibrillarin (Ab18380) antibodies were all from Abcam (Cambridge, UK).
Cells and Culture
HEK293 (immortalized human embryonic kidney cells), QZG (immortalized human liver cells), HepG2 (human liver carcinoma cells), and NIH3T3 (mouse fibroblasts) cells were cultured with DMEM supplemented with 10% fetal calf serum (FCS) with 5% CO2 at 37C.
IHC and Immunofluorescence
IHC with tissue specimens was performed using the Histostain-Plus SP kit (Zymed; South San Francisco, CA). Briefly, the sections were deparaffinized with xylene and rehydrated through gradient ethanol immersion. Endogenous peroxidase activity was quenched by 0.3% (v/v) hydrogen peroxide in methanol for 20 min, followed by three 5-min washes with PBS. Sections were then blocked with 10% (v/v) normal goat serum in PBS for 1 hr followed by overnight incubation at 4C with the anti-CIAPIN1 antibody, diluted 1:200, initial concentration 1.8 mg/ml, in PBS containing 3% (wt/vol) BSA. After three 5-min washes with PBS containing 0.02% (v/v) Tween-20 (PBST), sections were treated with biotinylated goat anti-mouse antibody for 20 min at room temperature, followed by three additional 5-min washes with PBST. Specimens were then incubated with streptavidin-avidin horse-radish peroxidase (HRP) for 20 min at room temperature, followed by repeated washes as described above. Reaction product was visualized with DAB at room temperature for 5 min. After being counterstained with hematoxylin for 30 sec and rinsed with tap water, sections were immediately dehydrated by sequential immersion in gradient ethanol and xylene and mounted with Permount (Fisher Scientific; Pittsburgh, PA) and coverslips. Images were obtained under a light microscope (BX51; Olympus, Tokyo, Japan) equipped with a DP70 digital camera.
For immunofluorescence, cells (both transfected and untransfected) grown on coverslides were immediately placed on ice, washed twice with chilled PBS, incubated for 5 min with chilled methanol and 30 sec with chilled acetone, and then dried at room temperature. Upon immunostaining, coverslips were incubated with the primary antibody (anti-CIAPIN1 MAb, 1:400 diluted and anti-6XHis MAb, 1:200 diluted) in PBST supplemented with 3% BSA at 4C overnight. After being rinsed three times for 10 min each in PBST, coverslips were incubated with TRITC-conjugated goat anti-mouse IgG (1:200 diluted in PBST plus 1% BSA; Beijing Zhongshan Biotechnology Co., Beijing, China) at room temperature for 1 hr and were then thoroughly rinsed again. Nuclei were stained with Hoechst 33,258. After being washed, cells were observed under a fluorescence microscope. Fluorescence images were taken under an Olympus BX51 microscope equipped with DP70 digital camera and the DPManager (DPController) software.
Plasmid Construction, Cell Transfection, and Anti-His Tag MAb Immunostaining
hCIAPIN1 coding region was obtained by PCR using the recombinant plasmid pUCm-T-hCIAPIN1 (constructed by our laboratory) as a template. Primers used in PCR were 5′-GAATTCATGGCAGATTTTGGGATC-3′ (forward) and 5′-GGGCCCGGCATCAAGATTGCTAC-3′ (reverse). EcoRI and ApaI sites (underlined) were introduced to the primers for convenience of subcloning. PCR product was isolated and cloned into pGEM-T easy vector and then subcloned into pcDNA 3.1/V5-His A, yielding recombinant plasmid pcDNA3.1/V5-His-hCIAPIN1 that was used to transfect HEK293 and NIH3T3 cells.
Transfection was carried out using Lipofectamine 2000 (Invitrogen; Carlsbad, CA) following the manufacturer's instructions. Briefly, 24 hr before transfection, cells (2 × 105 cells/well) were seeded into a six-well plate with sterile coverslips placed on the bottom of the wells. Upon transfection, the old medium was replaced with fresh serum-free DMEM, and the transfection complex was added. Eight hr later, the medium containing transfection complex was replaced with fresh DMEM supplemented with FCS. Forty eight hr after transfection, the coverslips were removed, fixed, and subjected to immunofluorescent staining as described above except that mouse anti-6XHis tag MAb was used as the primary antibody. Untransfected cells were processed just as described above to serve as controls.
Analytical Cell Fractionation
Cell fractionation and nucleolus isolation were carried out using the methods previously described (Muramatsu and Onishi 1978). When HepG2 cells grew to ∼80% confluence, the cell layer was washed with ice-cold PBS, pH 7.4, scraped off on ice, and collected by centrifugation at 500 × g for 5 min. Cells (∼1 × 108) were then washed three times with PBS, resuspended in 15 volumes of a hypotonic buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, and 1 mM MgCl2), and incubated on ice for 30 min. Cell lysis was performed by adding Nonidet P-40 to a final concentration of 0.3% and homogenization using a Dounce homogenizer with a tight pestle until >90% cells were broken and nuclei released. Dounced nuclei were centrifuged at 1200 × g for 5 min at 4C. The nuclear pellet was resuspended in 10 volumes of 0.25 M sucrose with 10 mM MgCl2. The supernatant fraction was harvested as the cytoplasm and stored for further analysis. Nuclei were purified by centrifugation at 1200 × g for 10 min through a 0.88 M sucrose cushion containing 0.05 mM MgCl2. Purified nuclei were resuspended in 10 volumes of 0.34 M sucrose containing 0.05 mM MgCl2 and sonicated on ice for several bursts of 30 sec with 5-min intervals in between. Nucleoli were then purified from the resulting homogenate by centrifugation at 2000 × g for 20 min through a 0.88 M sucrose cushion containing 0.05 mM MgCl2. The pellet contained the nucleoli, whereas the supernatant consisted of the nucleoplasmic fraction. Nucleoli were then washed by re-suspension in 500 μl of 0.34 M sucrose containing 0.05 mM MgCl2, followed by centrifugation at 2000 × g for 2 min at 4C. Samples of nuclei and nucleoli were observed under a phase-contrast microscope (CKX41; Olympus).
Cells, cytoplasm, nuclei, nucleoplasm, and nucleoli were all lysed with RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.1% SDS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.02% sodium azide, 100 μg/ml PMSF, 1 μg/ml aprotinin) for 30 min on ice. Concentration of total proteins was measured by the Bradford assay. For examination of the quality of cell fractionation, 100 μg (each lane) of protein was separated on a 15% polyacrylamide gel and stained with Coomassie Brilliant Blue. For Western blotting, 50 μg total proteins was loaded on each lane and separated on a 12% SDS-polyacrylamide gel and blotted onto nitrocellulose membrane (Millipore; Bedford, MA) using a semidry transfer system (BioRad; Hercules, CA). After being blocked in 5% non-fat milk at room temperature for 30 min, the membrane was incubated in the primary antibodies (anti-CIAPIN1 MAb, 1:400; anti-GAPDH, histone H1, and fibrillarin antibodies, 1:500; diluted in 2.5% non-fat milk-PBST) overnight. After incubation in HRP-conjugated goat anti-mouse IgG (1:2000; Beijing Zhongshan Biotechnology Co.) for 1 hr at room temperature and washed with PBST four times, the blot was detected with an enhanced chemiluminescent method (Amersham Pharmacia Biotech; Sunnyvale, CA).
Bioinformatics Analysis
PSORT II http://psort.nibb.ac.jp/) (Nakai and Horton 1999), SubLoc v1.0 http://www.bioinfo.tsinghua.edu.cn/SubLoc/) (Hua and Sun 2001), LOCSVMpsi http://bioinformatics.ustc.edu.cn/locsvmpsi/locsvmpsi.php/) (Xie et al. 2005), ESLPred http://www.imtech.res.in/raghava/eslpred/)(Bhasin and Raghava 2004), and HSLPred http://bioinformatics.uams.edu/raghava/hslpred/submit.html) (Garg et al. 2005) programs were used to predict the subcellular localization of CIAPIN1. In addition, we used NetNES 1.1 http://www.cbs.dtu.dk/services/NetNES/) to search for possible leucine-rich nuclear export signal (NES) in CIAPIN1 (la Cour et al. 2003,2004). Multiple sequence alignment was carried out with the ClustalW program (Thompson et al. 1994).
Results
IHC and Immunofluorescence
We first performed IHC staining to observe subcellular localization of CIAPIN1 in normal human tissues including fetal skeletal muscle, fetal colonic mucosa, adult brain, and adult gastric mucosa. As shown in Figure 1 under high magnification, immunostaining of both the cytoplasm and nucleus was clearly observed in the cells. Within the nucleus of some of these cells, structures similar to the nucleoli were identified. To achieve a higher resolution of the cell structure, we examined the intracellular distribution of CIAPIN1 in cultured cell lines including both human cells including normal cells HEK293 and QZG, hepatic cancer cells HepG2, and mouse fibroblast NIH3T3 cells with immunofluorescence. Results showed that although the overall intensity of CIAPIN1 immunostaining varied among cell types, distribution of the immunopositive signal diffused throughout the cells with the strongest staining in the nucleolus. Nonspecific immunostaining was strictly excluded by replacing anti-CIAPIN1 antibody with pre-immune mouse serum (Figure 2A).
Subcellular Localization by His-tagged CIAPIN1 Expression
We subsequently constructed the 6XHis-tagged CIAPIN1-expressing vector pcDNA3.1/V5-His-hCIAPIN1, transfected it into HEK293 and NIH3T3 cells, and immunofluorescently detected the subcellular distribution of exogenously expressed His-tagged hCIAPIN1 protein. As shown in Figure 2B, eukaryotic expression vector pcDNA3.1/V5-His mediated a high-level expression of His-tagged hCIAPIN1, resulting in a relatively strong overall immunofluorescent signal within cells. Distribution pattern of His-tagged hCIAPIN1 was identical to that of anti-CIAPIN1 immunostaining within untransfected cells.
Distribution of CIAPIN1 Among Cell Fractions
To verify the quality of HepG2 cell fractionation, we examined the purified nuclei and nucleoli using phase-contrast microscopy. Isolated nuclei and nucleoli had the typical characteristics (data not shown). We also performed SDS-PAGE to test the quality of the isolated cell fractions. The band patterns obtained using total cell lysate, cytoplasm, nuclei, nucleoplasm, and nucleoli were different (Figure 3A). These patterns were similar to those described by Scherl et al. (2002). To further confirm the quality of the cell fractionation, subcellular fractions were also detected by Western blotting with cytoplasmic marker GAPDH, nuclear marker histone H1, and nucleolar marker fibrillarin antibodies. As shown in Figure 3B, marker proteins were found in the expected fractions, indicating that the nuclei were well isolated.
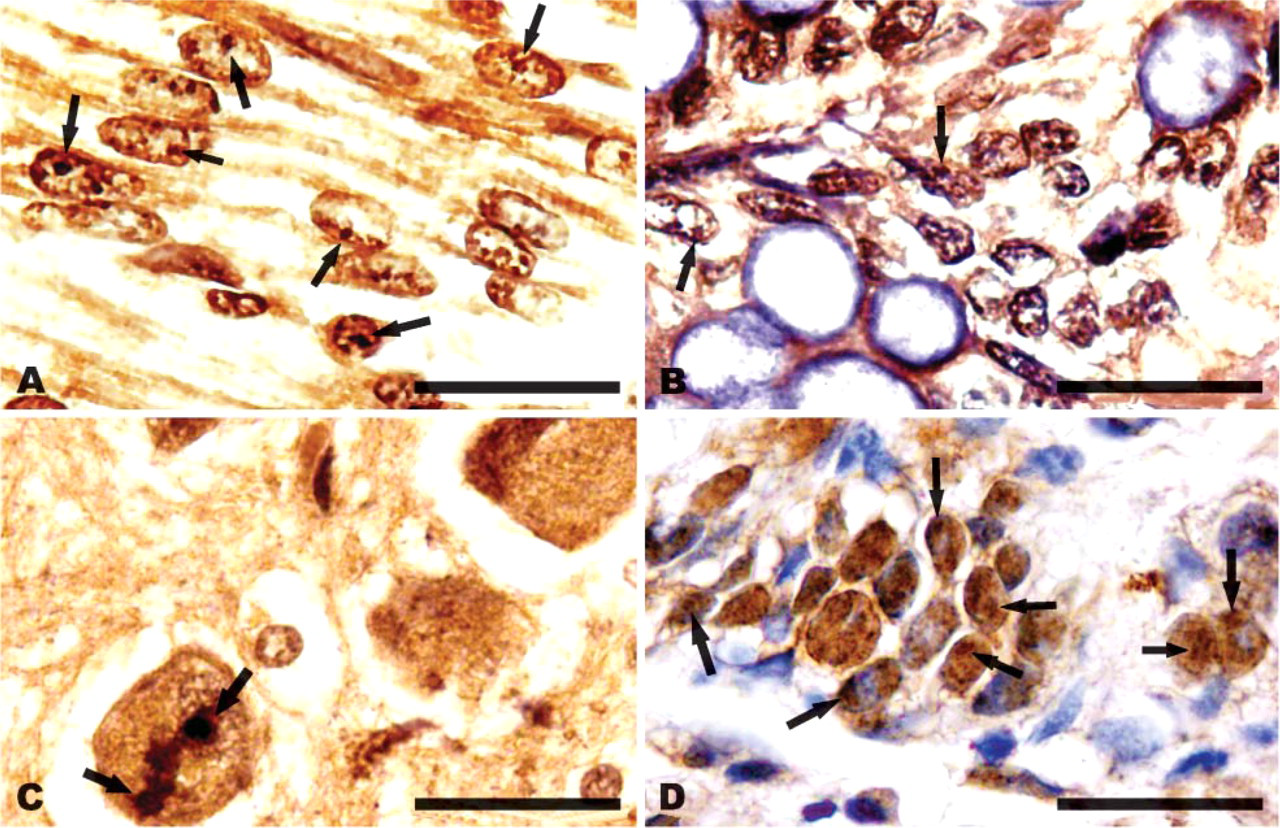
Immunostaining of CIAPIN1 in normal fetal and adult human tissues. (
Western blotting detection of the cell fractions with anti-CIAPIN1 MAb demonstrated a band of ∼39 kDa on each lane of total cell proteins, cytoplasm, nucleus, nucleoplasm, or nucleoli of HepG2 cells (Figure 3B), further confirming the subcellular localization demonstrated by the above means.
Bioinformatic Predictions Disclosed a Putative Nuclear Localization Signal (NLS) and a Putative NES
PSORT II, SubLoc v1.0, LOCSVMpsi, ESLPred, and HSLPred programs all predicted that CIAPIN1 is preferably localized in the nucleus (Table 1). PSORT II analysis disclosed a putative nuclear localization signal (NLS, KKRK, and amino acid residues 236-239 in hCIAPIN1 and 239-242 in mCIAPIN1) within the C terminus of CIAPIN1. Furthermore, the region surrounding this motif is rich in positively charged, basic amino acids arginine (R) and lysine (K). This is a prominent feature of nuclear protein.
Interestingly, the motif of (R/K)(R/K)X(R/K) residues has been identified to also be conserved in a number of nucleolar localization signal (NoLS) domains (Horke et al. 2004). We aligned the region around the motif KKRK of hCIAPIN1 with several other previously published nucleolar localization sequences of human proteins and found that the region between 324 and 372 amino acid residues had some similarity to these previously published NoLS sequences (including NoLS in La protein; telomerase reverse transcriptase; ribosomal proteins S7, L5, and S6; p14ARF; NOLP; ING-1; MDM2; IGF-1; and nucleolin) (Schmidt et al. 1995; Michael and Dreyfuss 1996; Annilo et al. 1998; Ueki et al. 1998; Lohrum et al. 2000; Rizos et al. 2000; Scott et al. 2001; Etheridge et al. 2002; Tan et al. 2002; Pellar and DiMario 2003; Horke et al. 2004), suggesting that this region might also serve as a NoLS.
In addition, NetNES 1.1 analysis revealed a putative leucine-rich NES within CIAPIN1 (amino acids 115-127, LCSALTLSGLVEV for hCIAPIN1 and amino acids 71-81, LCSALTLSGLV for mCIAPIN1).
Discussion
In our previous study, IHC staining of human and mouse tissues demonstrated that CIAPIN1 was localized to both the cytoplasm and the nuclei. To verify this finding, we further performed immunofluorescence, 6XHis-tagged fusion protein expression, and analytical cell fractionation in our present study. Results obtained by IHC and immunofluorescence, as well as 6XHis-tagged fusion protein expression, all demonstrated that both endogenously and exogenously expressed CIAPIN1 was localized to the cytoplasm and the nucleus and was accumulated in the nucleolus in both human and mouse cells. These results were further supported by analytical cell fractionation. In addition, bioinformatic prediction suggested that CIAPIN1 contains a NLS and a NES. Based on the above results, we postulate that CIAPIN1 may undergo a cytoplasm-nucleus-nucleolus translocation, which might be important for the functioning of CIAPIN1.
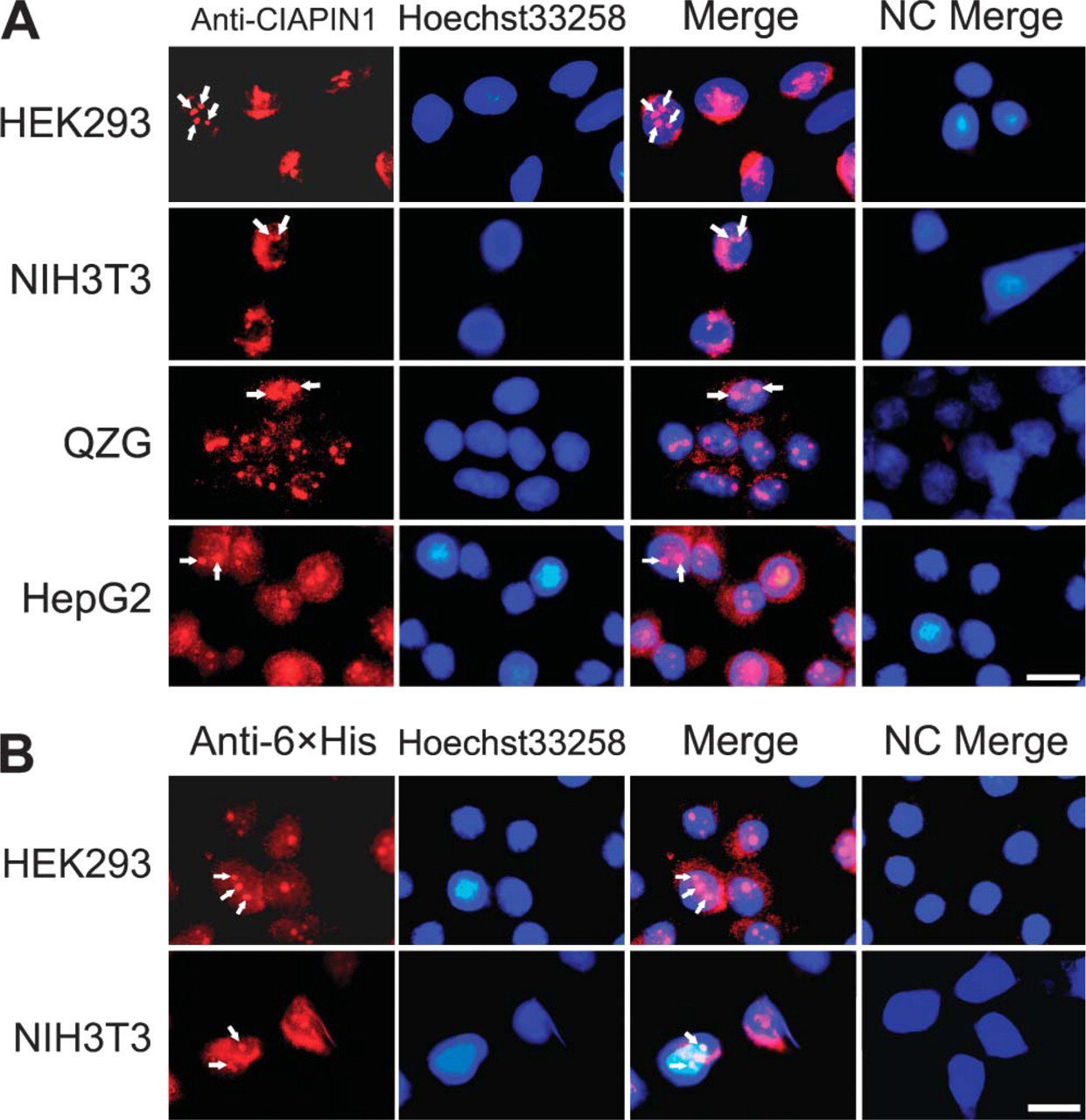
Immunofluorescent localization of CIAPIN1. (
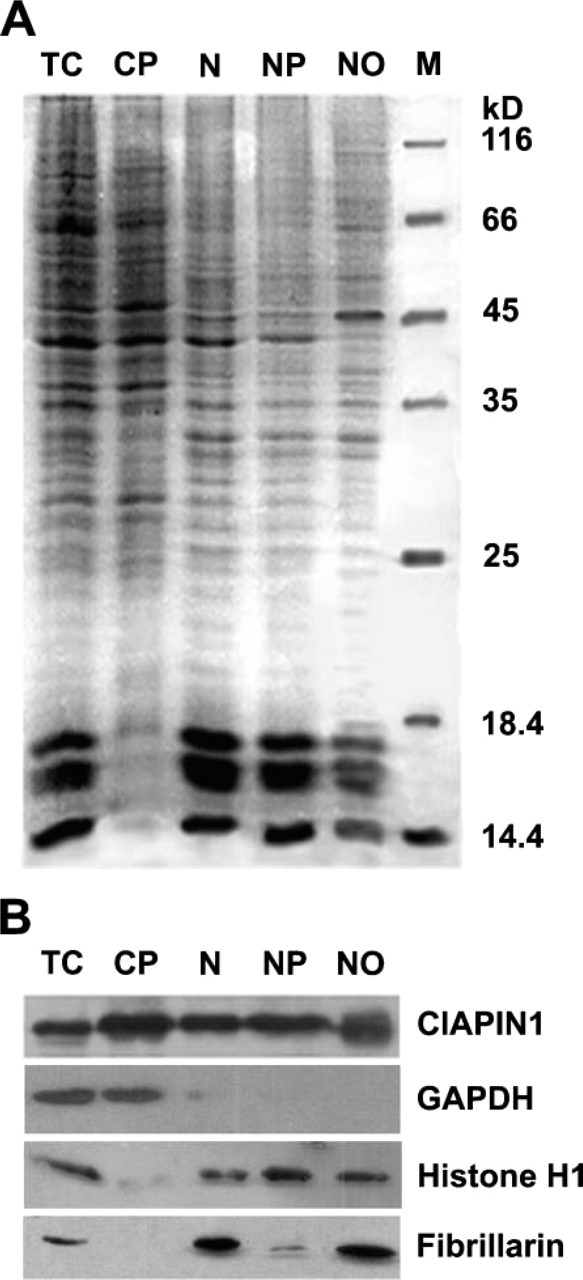
Distribution of hCIAPIN1 among HepG2 subcellular fractions. HepG2 cells were fractionated into cytoplasmic, nuclear, nucleoplasmic, and nucleolar fractions. (
Predicted subcellular localization of CIAPIN1
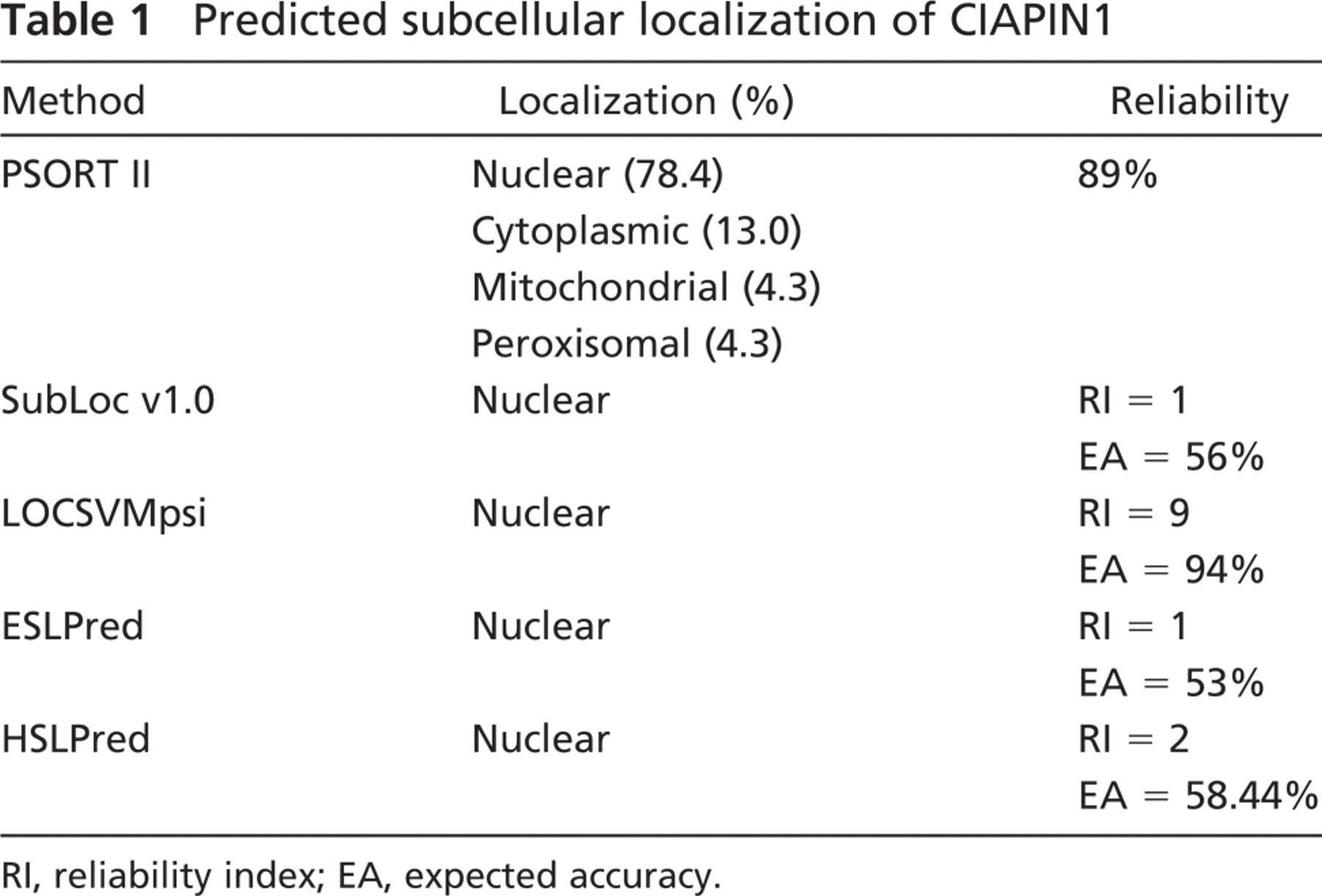
RI, reliability index; EA, expected accuracy.
Protein shuttling between nucleus and cytoplasm involves competitive interactions between NLS and NES or is mediated by a newly identified class of nucleocytoplasmic shuttling signals, which serves as both NLS and NES (Michael 2000). Most nuclear-cytoplasmic transport of proteins requires an NLS in the cargo and the nuclear transport receptors (karyopherins) that recognize NLS (Suntharalingam and Wente 2003). Although a strict consensus sequence for NLS has not been found among many nuclear proteins, NLS are typically short clusters of basic amino acids often preceded by an acidic amino acid or proline residue. However, a NLS may also consist of bipartite clusters of basic amino acids separated by a spacer region of approximately 10 amino acids, often flanked by a neutral or acidic amino acid. Furthermore, an impressive subset of NLS conforms to the consensus tetrapetide K-K/R-X-K/R, where × may be substituted by the amino acids K, R, V, P, and A but not by N (Chelsky et al. 1989). These basic amino acids are responsible for binding to the importin α receptor. Based on previously described NLS, a number of computational methods have been developed to predict putative NLS with rather high accuracy. Most of the cytoplasmic-nuclear shuttling proteins transport out of the nucleus via the interaction between NES and its receptor, CRM1/exportin 1/XPO1 (la Cour et al. 2004). Protein NES are hydrophobic-rich sequences with a characteristic spacing of leucine, isoleucine, valine, and/or phenylalanine. Leucine-rich NES consists of four or five hydrophobic residues within a region that is ∼10 amino acids in length. The widely accepted NES consensus is LX2,3[LIVFM]X2,3LX[LI] (la Cour et al. 2003). To date, ∼75 experimentally validated protein NES have been identified and compiled in the NESbase version 1.0 database (la Cour et al. 2003).
CIAPIN1 contains a KKRK tetrapeptide and the amino acid sequence neighboring this tetrapeptide is rich in basic amino acids, satisfying the criteria for a consensus NLS sequence. NetNES 1.1 program based on NESbase version 1.0 database disclosed a putative NES within both hCIAPIN1 and mCIAPIN1. These predicted results were consistent with and supported our experimentally verified subcellular localization of CIAPIN1.
A prominent feature of the subcellular localization of CIAPIN1 revealed in this study is its accumulation in the nucleolus. The subnuclear localization of proteins is either established because of specific signal sequences (e.g., NoLS) or is determined via interaction with specific molecules. Although analysis of the protein composition of the nucleolus does not lead to the identification of a general nucleolar targeting signal, it has been reported that the tetrapeptide R/K(R/K)X(R/K) motif appears as single or multiple copies in a region enriched with basic and hydrophilic amino acids and serves as the NoLS in a number of nucleolar proteins (Schmidt et al. 1995; Michael and Dreyfuss 1996; Annilo et al. 1998; Ueki et al. 1998; Lohrum et al. 2000; Rizos et al. 2000; Scott et al. 2001; Etheridge et al. 2002; Tan et al. 2002; Pellar and DiMario 2003; Horke et al. 2004). These amino acid residues tend to be externally oriented with respect to their structural position, thereby allowing for the interaction of nucleolar proteins with other molecules, which direct or retain the nucleolar proteins in the nucleolus. CIAPIN1 contains such a region overlapping the predicted NLS. This region might also be responsible for its nucleolar localization in addition to serving as a NLS, although further studies should be carried out to substantiate this proposal.
The major role of the nucleolus is in ribosome sub-unit biogenesis. In the nucleolus, which is considered a “cellular factory”, 28S, 18S, and 5.8S rRNAs are transcribed and, together with 5S rRNA, processed and assembled into the ribosome subunits. Recent studies have suggested that there may be additional functions for the nucleolus (Pederson 1998a; Olson et al. 2000). For example, it may also be a site for the biogenesis and/or maturation of other ribonucleoprotein machines, including the signal recognition particle (Politz et al. 2000), the spliceosomal small nuclear ribonucleoproteins (Lange and Gerbi 2000), and telomerase (Mitchell et al. 1999). The nucleolus may also participate in processing or exporting some mRNAs and tRNAs (Bertrand et al. 1998; Olson et al. 2000). Furthermore, an exclusive role of the nucleolus as a ribosome factory may not explain the recent discovery of viral, cell cycle regulatory, and tumor-related proteins and growth factors within this structure (Bouche et al. 1987; Henderson et al. 1995; Antoine et al. 1997; Garkavtsev et al. 1997; Galcheva-Gargova et al. 1998; Pederson 1998b; Visintin and Amon 2000; Schickling et al. 2001; Hiscox 2003; Arabi et al. 2005). The significance of localization of these non-classic nucleolar components in the nucleolus is not fully elucidated. Whereas some may be retained in the nucleolus to avoid interacting with their potential downstream partners until a specific cell cycle stage or metabolic status, others may be translocated into the nucleolus for regulating the structure and function of the nucleolus (Visintin and Amon 2000). This notion is supported by the findings that basic fibroblast growth factor and c-Myc, having been translocated into the nucleolus, exert their functions, at least in part, by activating RNA polymerase I transcription (Bouche et al. 1987; Arabi et al. 2005), whereas DEDD, a novel death effecter domain-containing protein, exerts its functions by activating caspase-6 and subsequently inhibiting RNA polymerase I transcription (Schickling et al. 2001). It is likely that nucleolar accumulation of CIAPIN1 falls into the above notion, although further investigations are required to clarify the pathway.
In conclusion, our preliminary study demonstrated that CIAPIN1 is localized in the cytoplasm and the nucleus and is accumulated in the nucleolus in both human and mouse cells. These findings suggest that CIAPIN1 may undergo a cytoplasm-nucleus-nucleolus translocation. Further studies are prompted to clarify the significance of the nuclear localization and the predominant nucleolar accumulation of CIAPIN1. Putative NLS and NES within CIAPIN1 also remain to be substantiated.
Footnotes
Acknowledgements
This work was supported by a grant from the Chinese National Foundation of Natural Sciences (No. 30471989).