
Abstract
To facilitate the immunological reaction of antibodies with antigens in fixed tissues, it is necessary to unmask or retrieve the antigens through pretreatment of the specimens. However, adjustment of heating-induced antigen retrieval is always required for different tissues and antigens. In this study, by using a low-power antigen-retrieval technique with appropriate dilution of antibodies, we successfully immunostained key antigens in pancreas such as insulin, PDX-1, glucagon, cytokeratin, and CD31, which have previously presented a particular challenge for investigators because of the rapid autodigestion and high nonspecific antibody binding in this tissue. Satisfactory results were obtained when immunohistochemistry and fluorescence in situ hybridization analysis were combined in the same slides.
Keywords
F
Following sacrifice, pancreas from adult immunodeficient (Nod/Scid/β2-microglobulin-null) (Nod/Scid/β2Mnull) mice (The Jackson Laboratory; Bar Harbor, ME) were dissected and fixed in 10% neutral-buffered formalin (pH 6.8-7.2) (Richard-Allan Scientific; Kalamazoo, MI) for 6 to 24 hr and then embedded in paraffin (Leica; Nussloch, Germany) and sectioned using a microtome (Leica). Five-μ-thick slides were dewaxed with 100% toluene (Sigma-Aldrich; St Louis, MO) and rehydrated. LAR was performed as follows: a maximum of eight slides were placed into a plastic jar (EMS; Hatfield, PA) filled with 275 ml, 1:100 Vector unmasking buffer (pH 6.0) according to manufacturer's instructions (Vector Laboratories; Burlingame, CA). The jar was located in the center of the microwave with lid off and was heated on a turnable plate in a microwave oven with multiple power settings (NN-S763WF, Genius Sensor 1350 W; Panasonic, Secaucus, NJ) at power level 4 (40%) for three cycles, 5 min for each cycle with 1-min break. one slide jar was processed each time. Although the temperature reached 100C in the jar, there was no bubbling or overflow and, thus, the buffer level in the jar remained unchanged in the whole process, allowing even treatment of all the slides in the jar. In contrast, when some slides were treated with conventional power level 10 (100%), buffer had to be replenished in each cycle due to the loss of fluid by overflow. The slides treated with low power and conventional power were cooled at room temperature on the bench for 30 min and then unmasking buffer was replaced with Tris-buffered saline containing 0.1% Tween-20 (TBST). Nonspecific binding was blocked with 250-300 μl, 100 mM, pH 7.5, TBST with 1% BSA and 5% normal donkey serum (IgG free, protease free; Jackson ImmunoResearch Laboratories, West Grove, PA) for at least 30 min until primary antibody was added. The proper working dilutions for each antibody were optimized for the system as shown in Table 1 to give the strongest specific antigen staining with the lowest nonspecific background.
Optimal conditions of immunohistochemistry analysis of murine pancreas with LAR
Published data (Wang et al. 2006).
1, Novus Biologicals; Littleton, CO; 2, Sigma; St Louis, MO; 3, Santa Cruz Biotechnology; Santa Cruz, CA; 4, Dako Cytomation; Carpinteria, CA; 5, Jackson ImmunoResearch Laboratories; West Grove, PA.
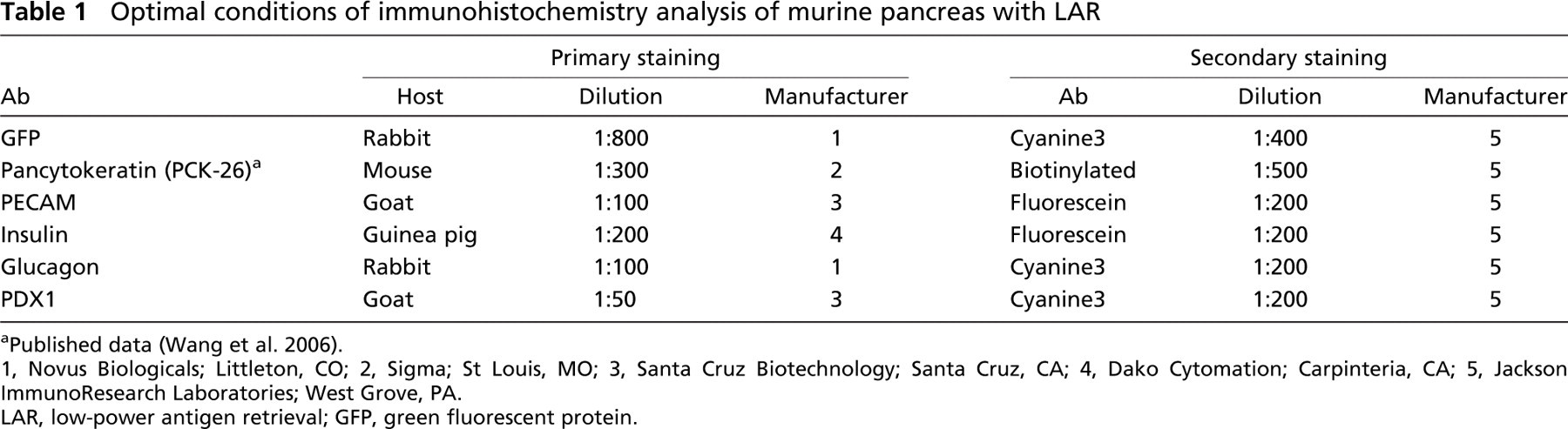
Specific staining of pancreatic cells in murine pancreas using low-power antigen-retrieval (LAR) protocol. Insulin+ β cells are shown in green and pancytokeratin (CK+) pancreatic duct epithelial cells in red (
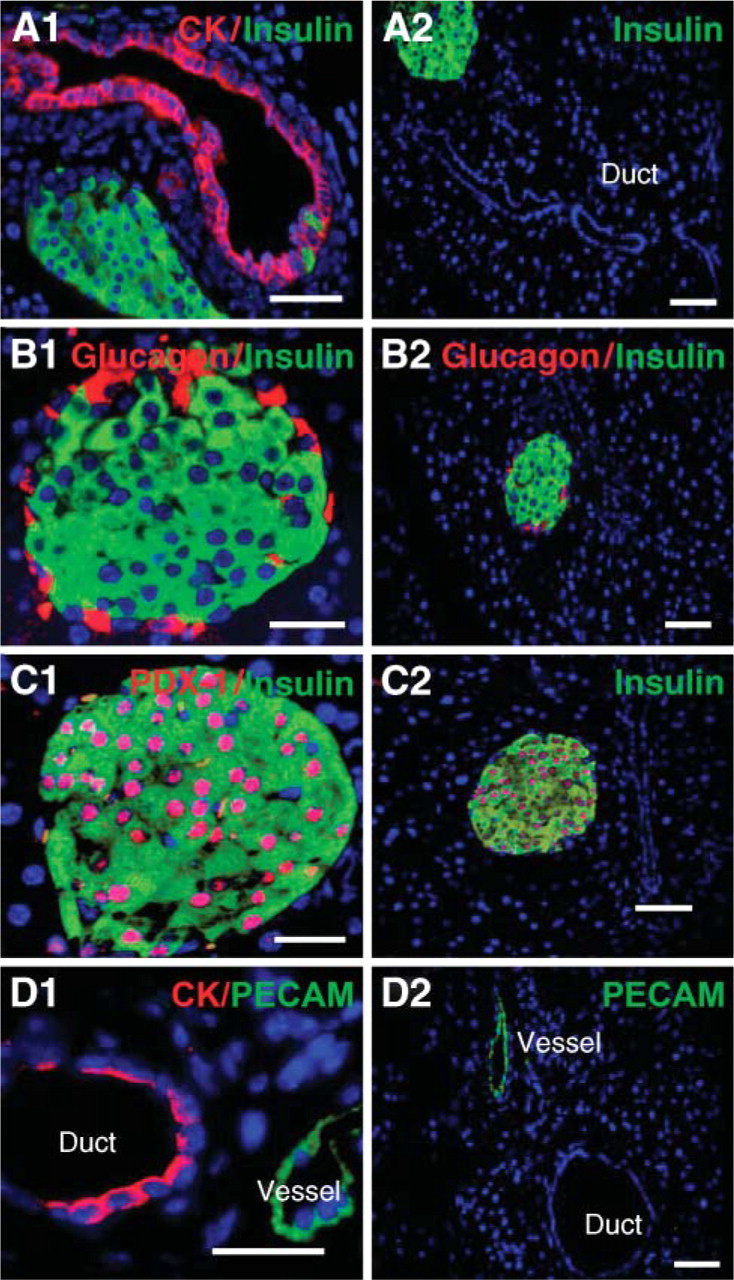
Comparison of PDX-1 expression in pancreas with low- and conventional-power AR treatment. Two consecutive pancreatic sections were treated with microwave oven under power 4 and power 10, respectively, for AR followed by identical dual staining and imaging procedure. PDX-1 signals (red) were greatly increased with LAR technique. Bar = 50 μm.
Images were viewed with a Leica DMRXA microscope (Bannockburn, IL) using a Plan Apo x20/1.0 or 1.25 numerical aperture, phase 3 differential interference contrast. Filter sets used were DAPI, chroma 31000, fluorescein 41001, Cy3,41007a (Chroma Technology; Rockingham, VT). The microscope was equipped with a LS300W ozone-free xenon arc lamp (Sutter Instrument; Novato, CA) coupled to the microscope with a liquid light guide. Images were acquired with an Applied Spectral Imaging Ltd. SkyVision-2/VDS-1300 12-bit digital camera from EasyFISH software (Migdal Ha'Emek, Israel). To compare the sensitivity and specificity of the signals after low- and conventional-power treatment, identical exposure time and intensity parameters were used.
Figure 1A demonstrates fluorescent signals of insulin for β cells of pancreatic islets and pancytokeratin (CK) for pancreatic duct epithelial cells on the same slides with no background. Interestingly, the successful costaining of insulin and CK enabled us to detect insulin-expressing cells (insulin+) within the ductal epithelial layer, considered by some investigators to be islet stem cells (Gu et al. 1994; Bonner-Weir 2000; Lechner and Habener 2003). Another component of the islets, the α cells that produce glucagons, could be identified in the peripheral areas of the islet surrounding the insulin+ cells after dual staining (Figure 1B). PDX-1 plays a key transcriptional role during the development of pancreas, and it is exclusively localized to β cells in the adult (Peers et al. 1994). Figure 1C shows the coexpression of insulin in cytoplasm and PDX-1 in nuclei of β cells of the pancreas, providing two markers for detecting β cells.
Blood vessel and pancreatic ducts both appear as hollow structures in the pancreas. When IHC staining with Abs against the epithelial marker CK and the endothelial marker PECAM (CD31) was applied to the pancreatic tissue, CK+ duct and CD31+ vessels were readily distinguished. There were no structures expressing both CK and CD31, demonstrating the specificity of the staining (Figure 1D). All staining showed high specificity and extremely low autofluorescence (Figures 1A2-1D2).
When dual staining of insulin and PDX-1 was performed in two consecutive sections with low- (power 4) and conventional-power (power 10) HIAR, respectively, PDX-1 signals were greatly improved with low-power technique under identical imaging control (Figure 2). The same results were found when dual staining of insulin and glucagon was performed (data not shown), indicating that high energy delivered by power 10 may kill antigens to some extent. In some cases, conventional power AR even caused failure of IHC staining (personal communication).
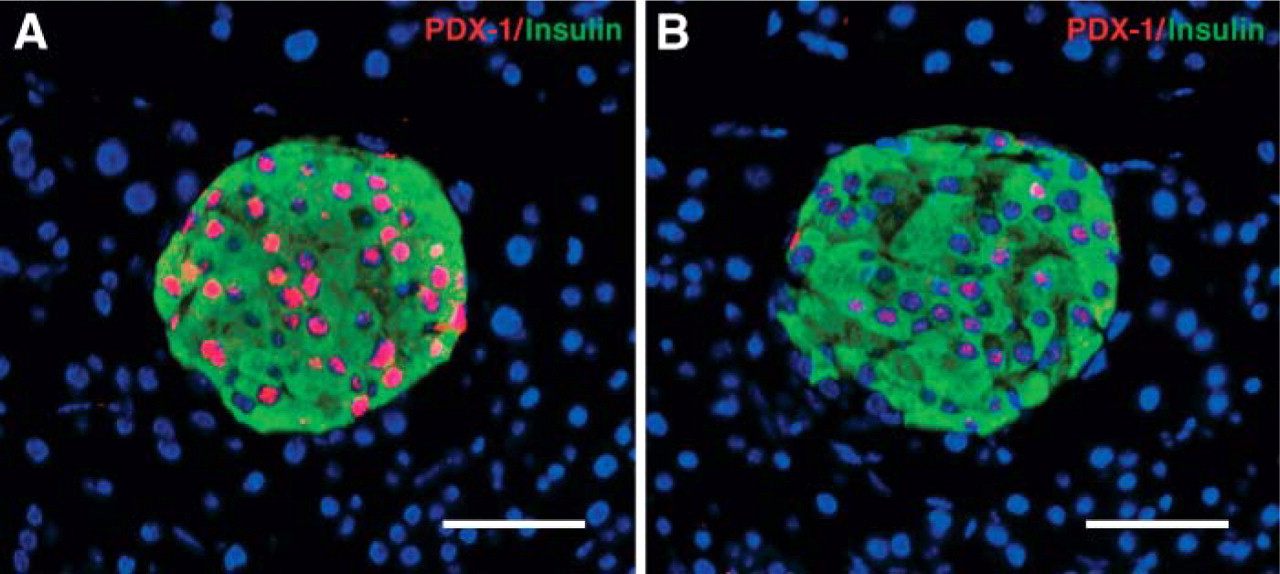
In the setting of transplantation where green fluorescent protein (GFP+) bone marrow cells from adult (10- to 12-week old) male mice transgenic for enhanced GFP gene (C57BL/6-TgN, ACEbEGFP 1Osb/J; The Jackson Laboratory) were used as donor, GFP detection was used to locate donor-derived cells in different tissues. using LAR, we successfully identified donor GFP+ cells in the pancreas (Figure 3) and other tissues from different mouse strains (as shown in Table 2). The application of LAR was extended to multiple antigen detection in different organs (Jodele et al. 2005; Kobayashi et al. 2005) (Table 2).
Colocalization of CK and green fluorescent protein (GFP) in donor-derived duct epithelial cells of recipient Nod/Scid/β2Mnull mice after bone marrow transplantation of GFP+ bone marrow fluorescence IHC staining of pancreas section from transplanted animal (
Applications of LAR with other tissues
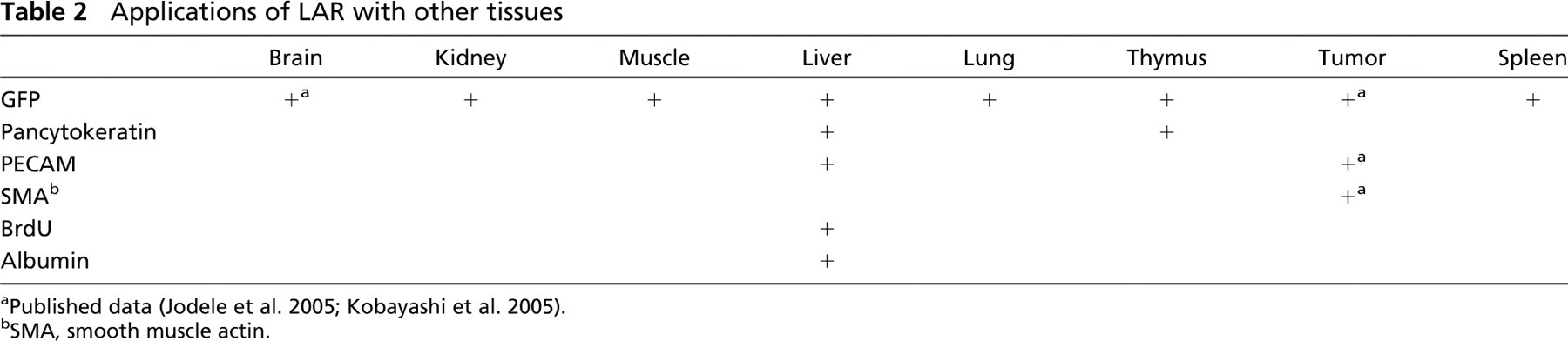
Published data (Jodele et al. 2005; Kobayashi et al. 2005).
SMA, smooth muscle actin.
It is often desirable to perform FISH and IHC on the same slide to see coexpression of specific chromosomes and protein, for example, to detect donor cells in sex-mismatched transplant settings. One of the primary technical concerns in these cases is that different procedures are required for FISH and IHC. We have found that unsatisfactory results of FISH staining can occur when IHC is performed prior to FISH staining. Vice versa, protein expression can be lost when FISH is processed first. In our studies, male pancreas was analyzed with IHC and FISH with the LAR technique. Slides were processed with LAR as described above, followed by digestion with 0.16% trypsin in diluent (Zymed; South San Francisco, CA) for 10 min at 37C and washing with TBST. After blocking, sections were incubated with primary Ab against the protein of interest, followed by incubation with biotinylated anti-mouse Ab, and then incubated with fluorescein (DTAF)-conjugated streptavidin (Jackson ImmunoResearch Laboratories). Post-fixation in 4% paraformaldehyde for 2 min, slides were sequentially dehydrated for 1 min in 70, 90, and 100% alcohol. One-μl Y-paint probe (Cy3) in 9-μl hybridization buffer (Cambio Ltd; Cambridge, UK) was applied on a 22 × 22-mm coverslip. Slides were sealed with rubber cement and denatured at 60C for 10 min and hybridized overnight at 37C in a humidified container. Slides were mounted with Vectashield (Vector Laboratories) medium containing DAPI after washing. Fluorescein and Cy3 filters were used to reveal protein and Y chromosomes, respectively.
It is well known that not all the Y chromosomes can be detected in male tissues due to incomplete sampling of entire nuclei during sectioning. At best, the efficiency of Y-chromosome detection in pancreas ranges from 60 to 80% with enzyme-mediated AR (Lechner et al. 2004) and in frozen section (Kodama et al. 2003). The LAR technique preserved protein expression well, e.g., insulin or CK with efficient detection of Y chromosomes. We observed 80-90% Y chromosomes of 2529 nuclei in the 5-μm sections of male pancreas with a background of 0.002% staining in negative (female pancreas) controls based on 2815 nuclei (Figure 4). LAR used here provided efficient antigen retrieval for successful immunostaining of a variety of antigens under a standardized condition with low background.
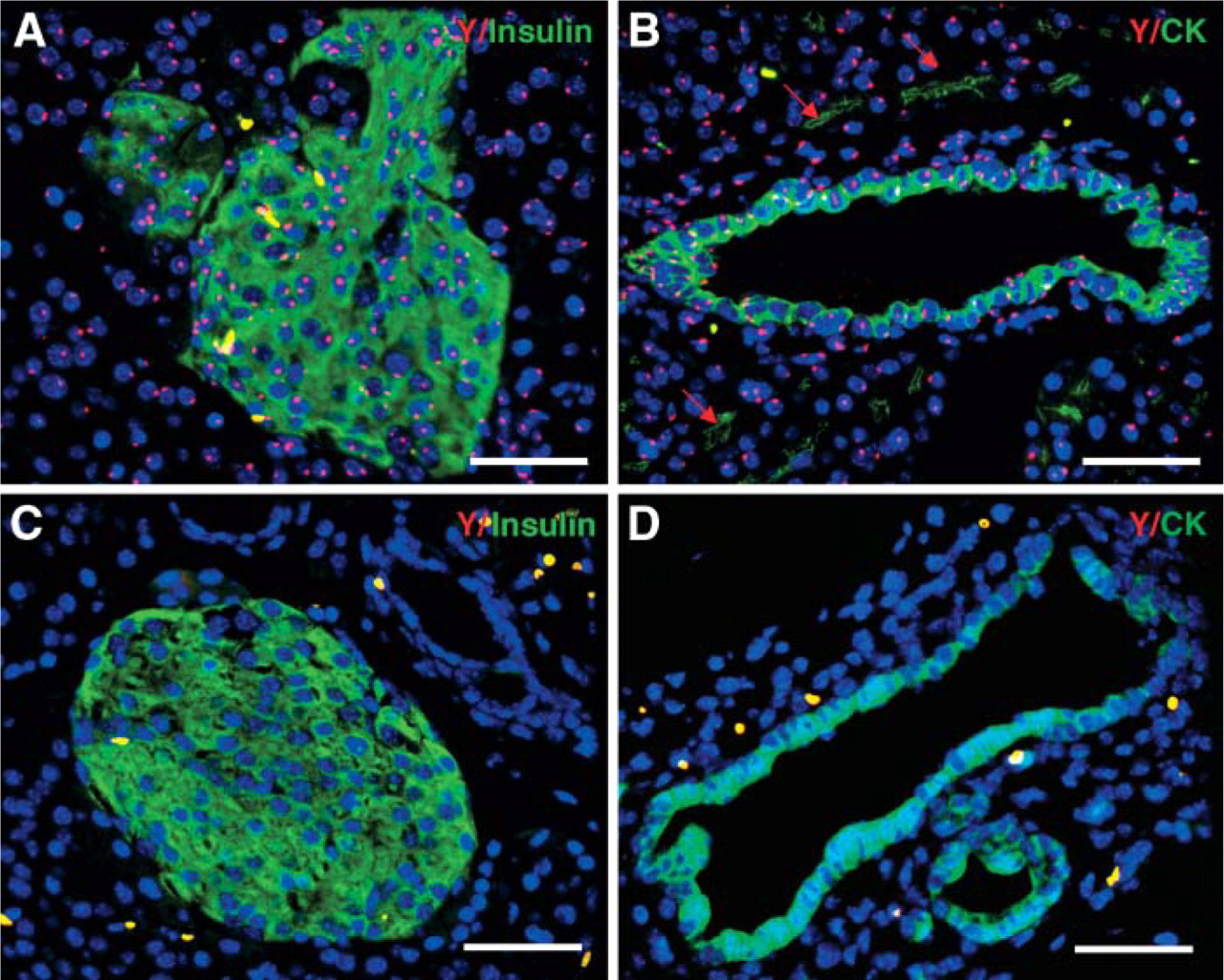
Efficient detection of Y chromosome in murine pancreas. Dual staining of insulin (green) (
Footnotes
Acknowledgements
This work was generously supported by grants from the Seaver Institute and NIH Grant (R01-DK-68719).
Fluorescence microscopy was performed in the Congressman Julian Dixon Cellular Imaging Core of the Saban Research Institute, Childrens Hospital Los Angeles.