
Abstract
Recently, in addition to DNA, RNA extracted from archival tissue specimens has become an invaluable source of material for molecular biological analysis. Successful amplification with PCR/RT-PCR is problematic when using amplicons of short size due to degradation of DNA or RNA. We established an improved method for efficient RT-PCR amplification of RNA extracted from archival formalin-fixed, paraffin-embedded tissue by the elimination of RNA modification and the restoration of RNA template activity. Namely, the preheating in citrate buffer (pH 4.0) of RNA extracted from long-term preserved tissue specimens resulted in significantly increased efficiency of RT-PCR.
Keywords
R
Extraction of DNA from formalin-fixed, paraffinembedded tissue for PCR analysis has been well documented (Jackson et al. 1990; Forsthoefel et al. 1992; Frank et al. 1996). On the other hand, RNA was first extracted from formalin-fixed, paraffin-embedded tissue for Northern and dot-blotting analysis (Rupp and Locker 1988). Subsequently, many reports were made about extraction of viral or human cellular RNA from archival samples and successful amplification of extracted RNA (Weizäcker et al. 1991; Bresters et al. 1992; Finke et al. 1993; Koopmans et al. 1993; Goldsworthy et al. 1999; Masuda et al. 1999; Korbler et al. 2003; Byers et al. 2004). Recent reports showed that RNA extracted from formalin-fixed, paraffin-embedded tissue samples was also available for quantitative analyses of gene expression (Lehmann and Kreipe 2001; Specht et al. 2001; Cohen et al. 2002; Kim et al. 2003; Cronin et al. 2004).
Nevertheless, persistent demands have been made for further improvement in RNA extraction from long-term preserved tissue samples whose RNA significantly degraded being chemically modified. The problems facing this goal include degradation of RNA in tissue due to delay before fixation, prolonged fixation, or long-term preservation after fixation (Bresters et al. 1994; Cronin et al. 2004), low efficiency of RNA extraction because of cross-linking with proteins (Finke et al. 1993; Park et al. 1996), and impaired reverse transcriptase reaction (Masuda et al. 1999) by formalin-induced modification (addition of mono-methylol to amino groups of four bases) of extracted RNA (Feldman 1973; Auerbach et al. 1977; Masuda et al. 1999). To overcome this problem, heat treatment of RNA prior to reverse transcription has been proposed. For example, it was reported that the chemical modification of all four bases of RNA by fixation in phosphate-buffered formalin can be reversed to some extent by incubation in TE buffer (pH 7.0) at 70C for 1 hr, resulting in restoration of template activity of RNA in RT-PCR (Masuda et al. 1999).
While performing molecular analyses on cancer tissue specimens taken from atomic bomb survivors and that were stored for several decades (up to 50 years), we frequently encountered archival unbuffered formalin-fixed, paraffin-embedded specimens that were difficult to use for RT-PCR analysis. We found that significant degradation and chemical modification of RNA greatly affected RT-PCR amplification.
Removal of chemical modification from bases of RNA as well as significant reduction of amplicon size may be crucial for the enhanced availability of limited archival unbuffered formalin-fixed, paraffin-embedded tissue samples. Therefore, we examined whether the preheating of RNA extracted from archival unbuffered formalin-fixed, paraffin-embedded tissues enhances the efficiency of RT-PCR, along with determining optimal conditions for the RNA preheating. Application of this preheating technique to retrospective research is expected to enhance the availability of archival unbuffered formalin-fixed, paraffin-embedded tissue specimens for molecular analysis that have been stored for several decades and have functioned as a source for histological evaluation, to allow better understanding of molecular characteristics of various diseases.
Materials and Methods
Tissue
Five archival unbuffered formalin-fixed, paraffin-embedded thyroid cancer tissue samples for in-house control were used in this study. All samples were preserved at room temperature for 19 to 21 years. After deparaffinization of 5-μm sections by Hemo-De (Fujisawa Yakuhin Kogyo; Osaka, Japan) and staining with methylgreen (Sigma-Aldrich; St. Louis, MO), cancerous regions (~2-3 × 3 mm) were isolated using a laser microdissection system (Leica AS LMD; Wetzlar, Germany). All cancerous regions microdissected from six to eight successive tissue sections were combined for RNA extraction.
RNA Extraction and Measurement
RNA was isolated from microdissected tissue using the High Pure RNA Paraffin Kit according to the manufacturers instructions (Roche Diagnostics; Basel, Switzerland), with some modifications. Briefly, microdissected tissue was digested with proteinase K at 55C overnight, followed by DNase I treatment. After the lysate was purified by High Pure filter, RNA was eluted twice with 100 μl of RNase-free water. RNA was then precipitated by ethanol in the presence of 2 μl of ethachinmate (Nippon Gene; Tokyo, Japan) as a carrier and resuspended in 30 μl of RNase-free water. The concentration of RNA was measured by absorption at 260 nm with a spectrophotometer (Gene Spec III; Hitachi, Tokyo, Japan). The quality of extracted RNA was measured by electrophoresis on 1.5% or 3.0% native agarose gel and 2.5% formaldehyde agarose gel.
Heat Treatment of RNA
Approximately 150 ng of total RNA was heated in 250 μl of 10 mM citrate buffer with various pH values ranging from 3 to 6.5 at 70C for a number of different time periods. Preheating in 250 μl of 10 mM sodium borate with various pH values (pH 6.5-10) or 10 mM TE buffer (pH 7.0, 7.5, and 8.0) was similarly carried out. After preheating, RNA was precipitated by ethanol in the presence of ethachinmate as carrier and dissolved in RNase-free water to arrive at a final concentration of 10 ng/μl.
cDNA Synthesis
One hundred ng of total RNA and 50 pmol/μl of random primers (9 mer) were heated in 11 μl of RNase-free water at 65C for 10 min and chilled in ice water. A mixture consisting of 4 μl of 5 × RT buffer, 2 μl of 20 mM DTT, 1 μ of 10 mM dNTPs, and 1 μl of RNase Inhibitor (20 U/μl; Takara, Tokyo, Japan) was added to RNA solution and incubated at room temperature for 5 min. After addition of 1 μl of ReverTra Ace (100 U/μl; Toyobo, Osaka, Japan), a reaction mixture was incubated at 42C for 60 min and at 70C for 15 min.
Detection of Expression of Breakpoint Cluster Region (BCR) and N-ras Genes by RT-PCR
RT-PCR was performed in a 25-μl volume containing 1 × PCR buffer, 200 μM each of dATP, dCTP, dGTP, and dTTP, 1.5-3.0 mM MgCl2, 0.4 μM of each specific primer, 0.5 U Platinum Taq DNA polymerase (Invitrogen; Carlsbad, CA), and 2 μl of cDNA from the previous RT reaction. Primary denaturation (95C for 3 min) and final extension (72C for 5 min) were the same for each RT-PCR reaction, all of which were subjected to 40 cycles of amplification consisting of 95C for 30 sec, 55-60C for 30 sec and 72C for 30-45 sec for BCR, and 95C for 30 sec, 52-55C for 30 sec and 72C for 30-45 sec for N-ras. For positive and negative controls of RT-PCR, cDNA derived from human thyroid cancer cell line (8505C) and H2O were used as templates, respectively. Five μl of the reaction mixture was run on an 8% acrylamide gel and visualized with ethidium bromide. In each experiment, it was confirmed that all target bands were the real ones by digestion of restriction enzyme, which existed within each amplified target fragment. Since all the primer sets used were designed to locate in two different exons, no amplification of RNA without RT was observed. The effects of preheating on RT-PCR efficiency were examined by amplifying the fragments of different sizes in the BCR gene (eight different sizes: 61 bp, 94 bp, 127 bp, 152 bp, 175 bp, 222 bp, 250 bp, and 275 bp) and the N-ras gene (seven different sizes: 61 bp, 98 bp, 121 bp, 148 bp, 199 bp, 221 bp, and 250 bp). The semiquantification of each PCR product was made by measurement of the intensity of each band using Kodak (Tokyo, Japan) 1D Image Analysis Software.
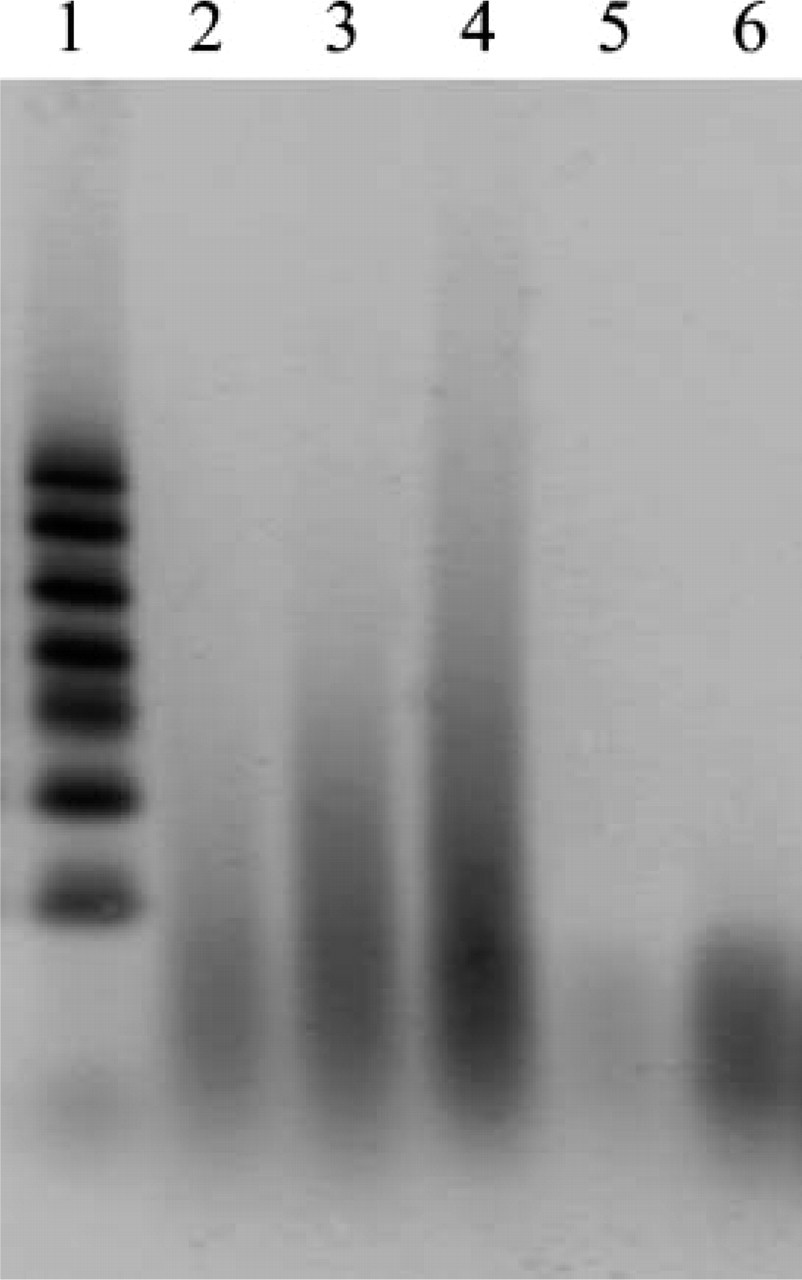
Total RNA extracted from archival unbuffered formalin-fixed, paraffin-embedded thyroid cancer tissue. The extracted RNA was electrophoresed on 3% native agarose gel. Lanes 2-4 contain RNA from three different archival samples. Lane 1, 100-nucleotide RNA marker; Lanes 5 and 6, 70 base-synthesized nucleotides (25 ng and 100 ng/lane, respectively).
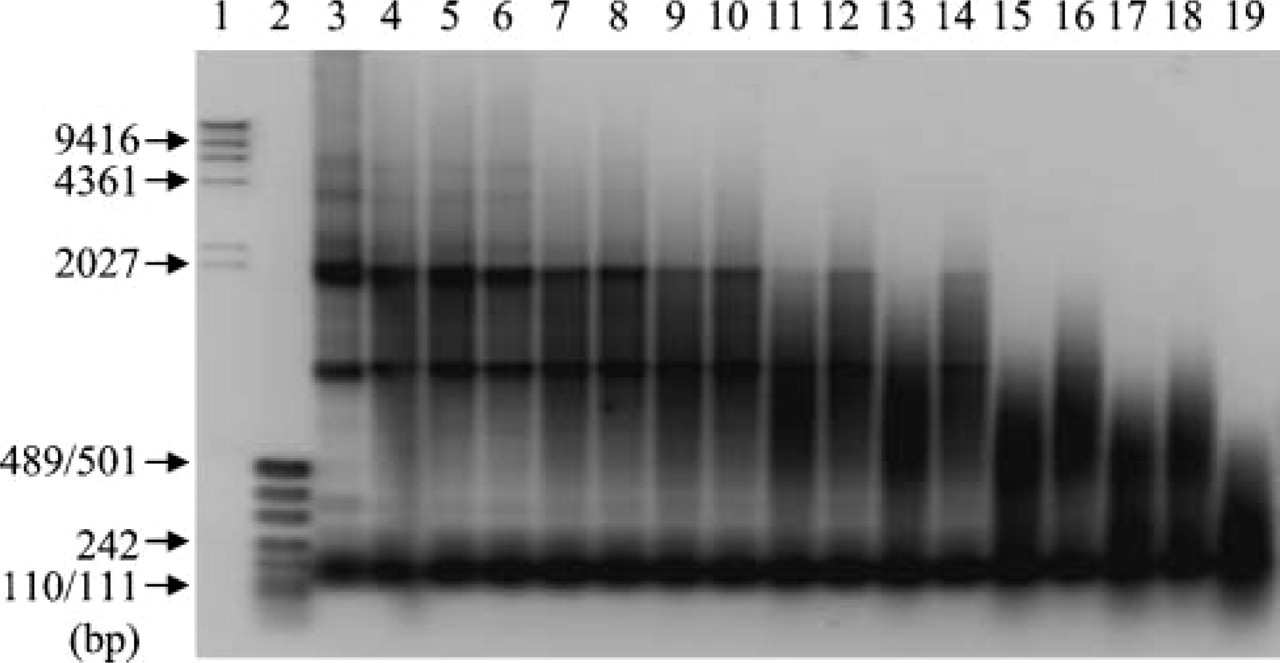
Effect of preheating temperature on RNA integrity. Two μg of total RNA extracted from frozen human thyroid cancer cell line were heated for 15 min or 30 min at various temperatures (Lanes 4 and 5, 60C; Lanes 6 and 7, 65C; Lanes 8 and 9, 70C; Lanes 10 and 11, 75C; Lanes 12 and 13, 80C; Lanes 14 and 15, 85C; Lanes 16 and 17, 90C; Lanes 18 and 19, 95C). Lane 3, no heating; Lane 1, λ HindIII DNA marker; Lane 2, pUC19-MspI digest for DNA size marker. Electrophoresis was done on 1.5% native agarose gel. Fifteen min heating is for Lanes 4, 6, 8, 10, 12, 14, 16, and 18. Thirty min heating is for Lanes 5, 7, 9, 11, 13, 15, 17, and 19.
Results
RNA Extracted from Unbuffered Formalin-fixed, Paraffin-embedded Tissue
We extracted RNA from five different archival unbuffered formalin-fixed, paraffin-embedded thyroid cancer tissue specimens, as described in Materials and Methods. An image of electrophoresis of these RNA is shown in Figure 1. They appeared as smears on agarose gel with no ribosomal bands observed in any of the samples. The range of smeared RNA differed slightly among five archival tissue specimens. A majority of the smeared RNA sample used for determination of the conditions for preheating of RNA ranged from ~70 to 100 bases (Figure 1, Lane 2). Other RNA samples showing better efficiency of RT-PCR amplification than the previous one ranged from ~70 to 200-300 bases (Figure 1, Lanes 3 and 4). The size of RNA from the remaining two tissue samples was the intermediate among the other three samples.
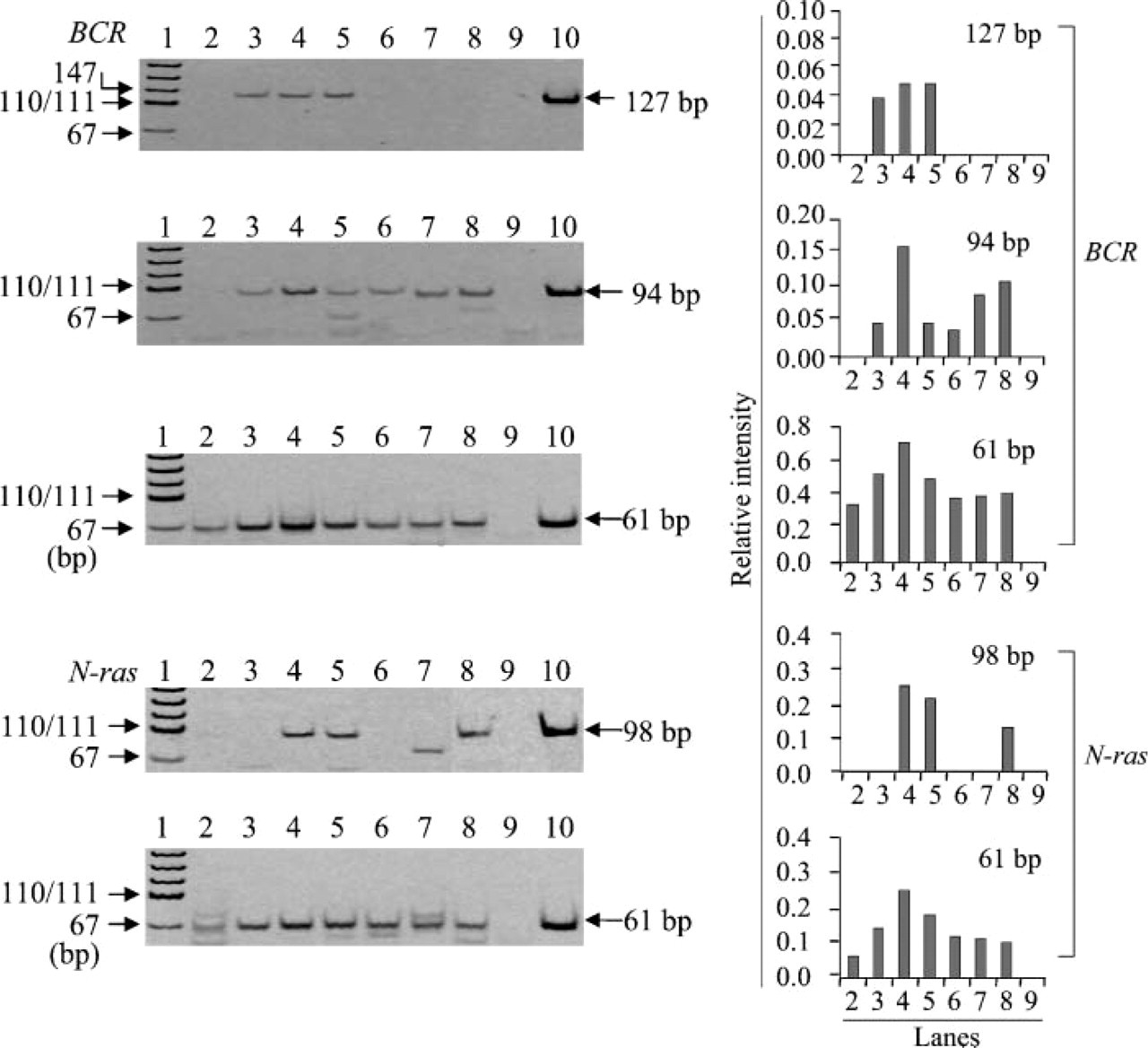
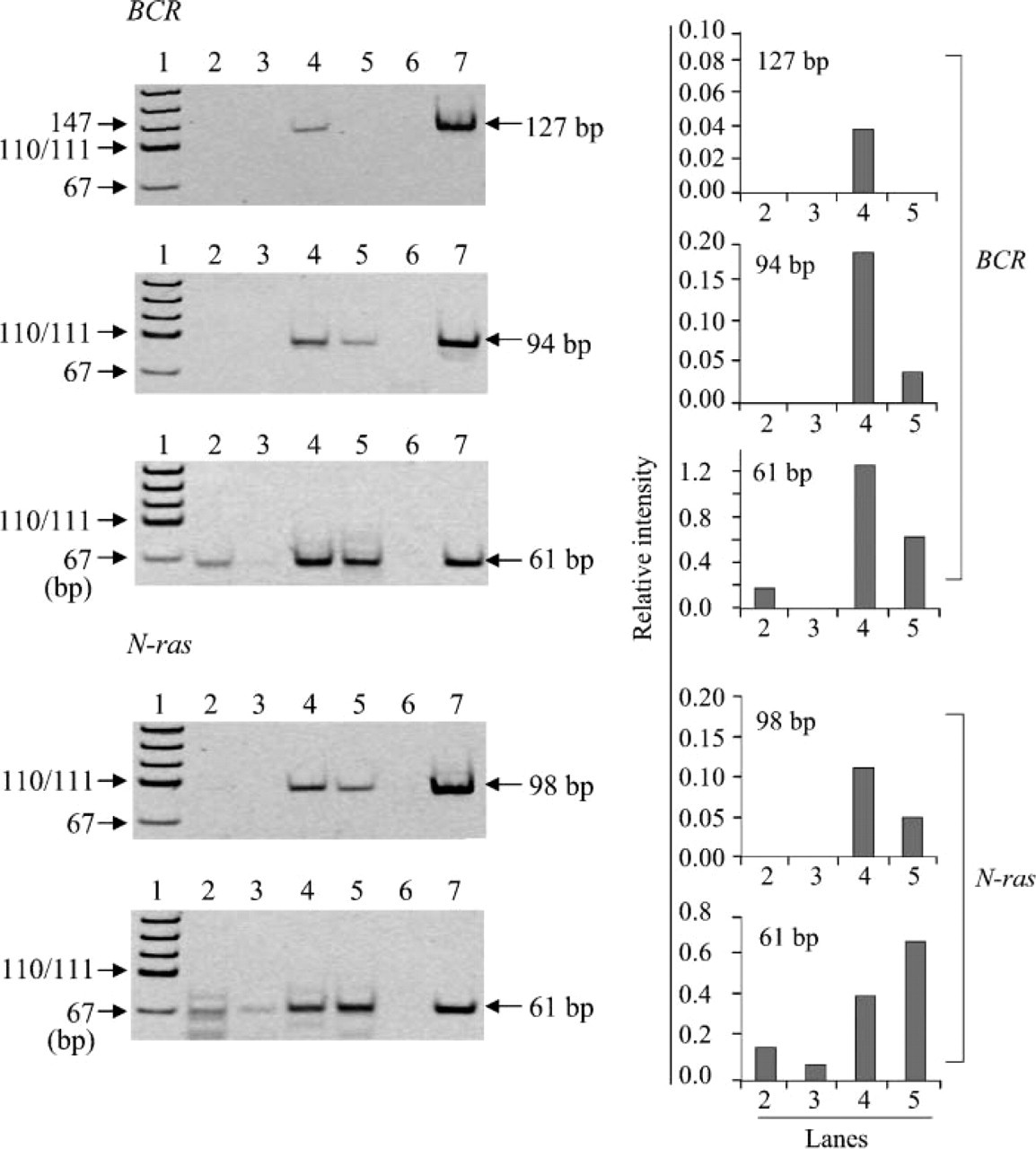
Effect of pH on RT-PCR amplification of RNA. Efficiency of RT-PCR amplification of RNA preheated at 70C for 30 min in various pHs (Lane 3, pH 3.0; Lane 4, pH 4.0; Lane 5, pH 5.0; Lane 6, pH 6.0; Lane 7, pH 6.5) was measured by amplifying 61-, 94-, and 127-bp fragments in the BCR gene and 61- and 98-bp fragments in the N-ras gene. Lane 2, no preheating; Lane 8, preheated with TE (pH 7.0) for 30 min; Lane 9, negative control; Lane 10, positive control; Lane 1, pUC19-MspI digest for DNA size marker. The bands different from the position shown by arrow indicate the extra bands. This is the same in Figures 2-5. Bar graphs at right indicate the relative intensity of each target band when intensity of positive control is assumed to be 1.0. The numbers on the horizontal axis correspond to the number of lanes in the left electrophoresis. This labeling is also used in Figures 3-5.
Temperature in Preheating of RNA
At first we tested the effects of incubation at different temperatures on RNA stability using intact RNA prepared from human thyroid cancer cell lines, because it is hard to evaluate the preheating effect using already degraded RNA extracted from formalin-fixed, paraffin-embedded tissue specimens (Figure 2). Preheating of RNA at >80C for 30 min in H2O resulted in vigorous degradation. Considering our result and a report by Masuda et al. (1999), we set preheating temperature at 70C in this study.
Effects of pH in Preheating of RNA on RT-PCR Amplification
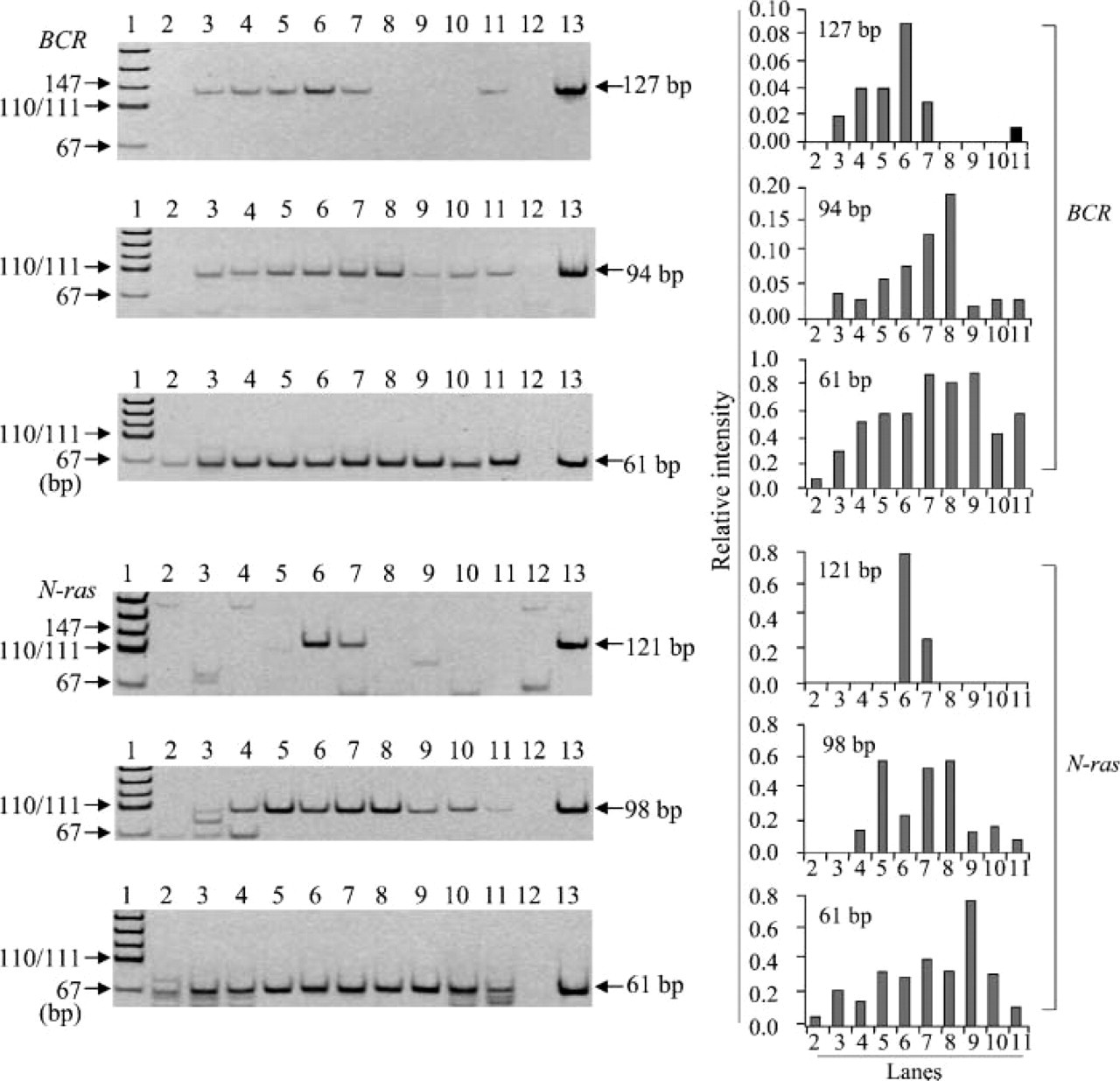
Using the RNA with the worst efficiency of RT-PCR amplification among the five archival tissue samples, we examined the effects of preheating on RT-PCR efficiency by amplifying the fragments of different sizes in the BCR gene and the N-ras gene (Figure 3). There were 61-bp fragments in the BCR and N-ras genes detected in the RNA that were not undergoing preheating, although the intensity of the bands was weak compared with that in the RNA with preheating (Figure 3). Preheating of RNA in 10 mM citrate buffer at pH 3-5 at 70C for 30 min made amplification of 94- and 127-bp fragments possible in the BCR gene and 98-bp fragments in the N-ras gene. These same fragments could not be amplified by RT-PCR without undergoing preheating (Figure 3). Preheating of RNA in citrate buffer with pH ~4 was the most effective method in the RT-PCR amplification of the BCR and N-ras genes. To determine optimal pH, we further investigated in detail the effects of pH on RT-PCR amplification using preheated RNA. As shown in Figure 4, RNA treated with ~pH 4.0 showed the most efficient RT-PCR amplification of both the BCR and N-ras genes among citrate buffers with different pH values ranging from 3 to 5 and TE (pH 7.0). We also examined the effect of pH range (6.5-10) on RT-PCR amplification using 10 mM sodium borate solution and TE. Among buffers ranging from pH 7.0 to pH 8.0 (TE buffers with pH 7.0, 7.5, and 8.0 and borate buffer with 8.0), little difference was found in the effect of preheating with these buffers at a concentration of 10 mM (data not shown). The effect of preheating decreased with increased pH, and adverse effect was observed in RNA treated with pH 9.0 or 10.0 (data not shown). Little difference was found in efficiency of RT-PCR amplification enhanced by preheating of RNA between citrate buffer (pH 6.5) and sodium borate (pH 6.5) (data not shown).
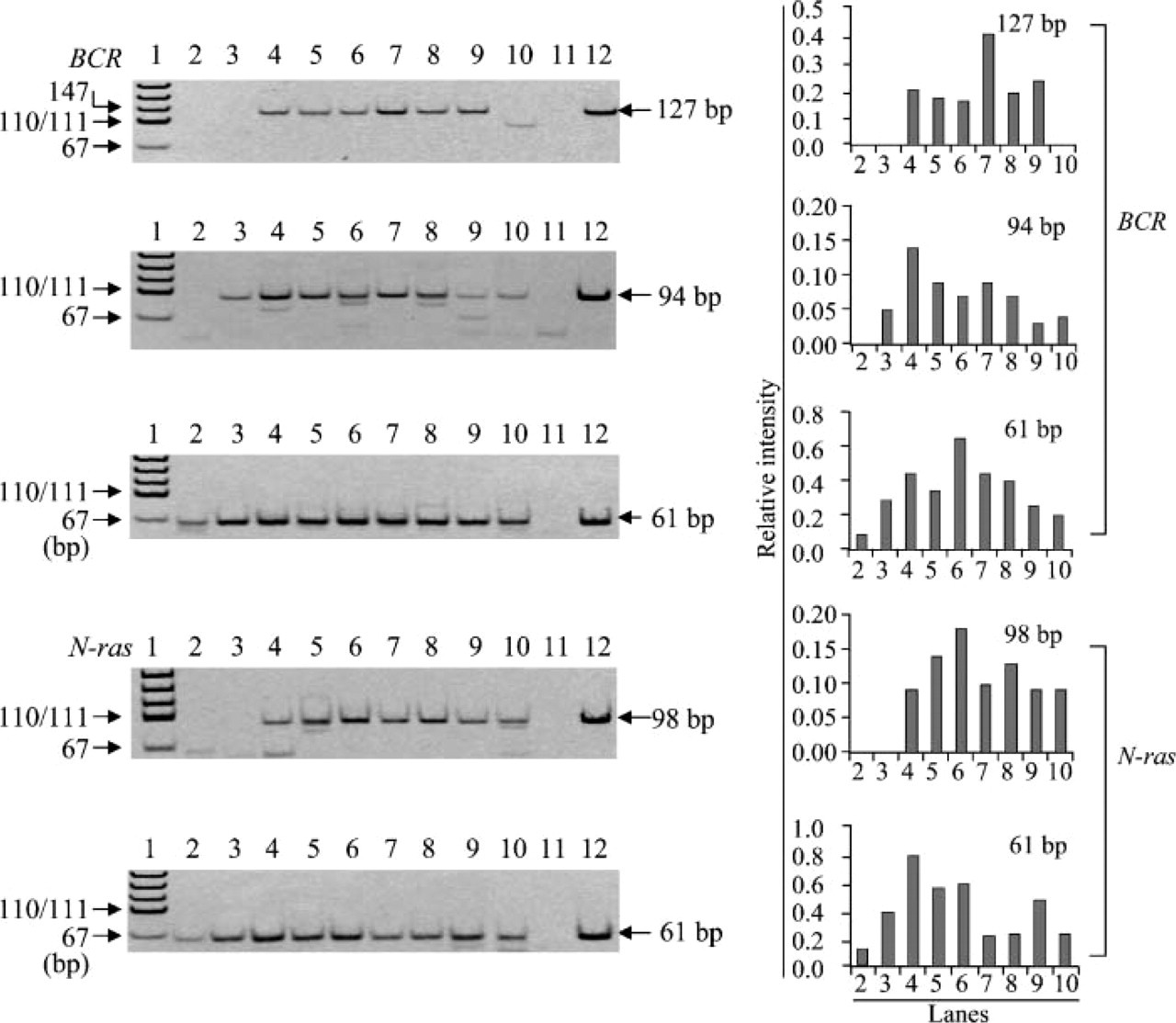
Detailed analysis of pH of citrate buffer ranging from 3.0 to 5.0 for optimization of preheating condition. RNA was preheated in citrate buffer (Lane 3, pH 3.0; Lane 4, pH 3.5; Lane 5, pH 3.7; Lane 6, pH 4.0; Lane 7, pH 4.25; Lane 8, pH 4.5; Lane 9, pH 5.0) at 70C for 30 min before cDNA was synthesized. RT-PCR amplification of BCR and N-ras mRNAs was carried out to determine the optimal pH for preheating of RNA. Lane 2, no preheating; Lane 10, preheated with TE (pH 7.0) for 30 min; Lane 11, negative control; Lane 12, positive control; Lane 1, pUC19-MspI digest for DNA size marker.
Effects of Buffer Concentration in Preheating of RNA on RT-PCR Amplification
We examined the effects of different buffer concentrations on RT-PCR amplification. In both BCR and N-ras target genes, when RNA was heated with 50 mM citrate buffer at 70C, even 61-bp fragments were hard to detect. There was a lower efficiency of RT-PCR amplification than amplification without preheating (Figure 5). On the other hand, treatment with 10 mM citrate buffer resulted in the greatest efficiency of RT-PCR amplification among concentrations investigated (Figure 5).
Effects of Preheating Time of RNA on RT-PCR Amplification
We examined the effects of preheating time on RT-PCR amplification. As shown in Figure 6, preheating in citrate buffer (pH 4.0) at 70C for 45 min was the most effective method for RT-PCR amplification. Thus, the efficiency of RT-PCR amplification depends on pH, concentration of buffer, and incubation time. In the other four archival thyroid tissue samples, we also examined whether preheating of RNA in citrate buffer (pH 4.0) at 70C for 45 min resulted in enhanced efficiency of RT-PCR amplification. We found that the longer fragments in the preheated RNA could be amplified in all cases compared with the non-treated RNA, although the amplified fragment sizes differed among these thyroid tissue samples (Figure 7). Increased efficiency of RT-PCR amplification, therefore, was observed in all five unbuffered formalin-fixed, paraffin-embedded thyroid tissue samples through the preheating of RNA in citrate buffer (pH 4.0).
Discussion
Multiple papers have reported improvement in efficiency of RNA extraction from buffered formalin-fixed, paraffin-embedded tissue. Few reports, however, have reported on elimination of modification induced by buffered formalin fixation, although RT-PCR amplification of RNA extracted from archival formalin-fixed, paraffin-embedded tissue was hindered not only by degradation of RNA but also by modification of RNA bases by formalin. Preheating of RNA in TE buffer (pH 7.0) restored the template activity of RNA extracted from buffered formalin-fixed tissue where clear bands of 18S and 28S rRNA were still detected with partial degradation (Masuda et al. 1999). The reaction between formaldehyde and nucleotide monomers takes place in two steps. Primary reaction is to form labile methylol-derivatives by addition of formaldehyde group to NH-group of bases. Secondary slow reaction is to give rise to stable methylene derivatives in only amino purines (Feldman 1973; Auerbach et al. 1977). The reaction between formaldehyde and RNA is also thought to occur in the same manner. In fact, matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) analysis has indicated that all four bases of RNA treated with buffered formalin were modified mainly by addition of mono-methylol groups (Masuda et al. 1999). Modification of nucleotides by addition of methylol groups is a reversible reaction: heating of RNA with 10 mM TE buffer (pH 7.0) at 70C results in removal of methylol derivatives from bases (Masuda et al. 1999).
Effect of concentration of citrate buffer on RT-PCR amplification of RNA isolated from archival thyroid tissues. RT-PCR amplification of RNA preheated at 70C for 30 min in various concentrations of citrate buffer with pH4.0 (Lane 3, 50 mM; Lane 4,10 mM; Lane 5, 2 mM) was performed on three different sizes of fragments in the BCR gene and two different sizes of fragments in the N-ras gene. Lane 2, no preheating; Lane 6, negative control; Lane 7, positive control; Lane 1, pUC19-MspI digest for DNA size marker.
Effect of preheating time on RT-PCR amplification. RT-PCR amplification of RNA preheated at 70C in citrate buffer (pH 4.0) for various preheating times (Lane 2, 0 min; Lane 3, 10 min; Lane 4, 20 min; Lane 5, 30 min; Lane 6, 45 min; Lane 7, 60 min; Lane 8, 90 min; Lane 9, 120 min) was done for different lengths of fragments in BCR and N-ras. Lanes 10 and 11, preheated with TE for 30 min and 60 min; Lane 12, negative control; Lane 13, positive control; Lane 1, pUC19-MspI digest.
Our results indicate that efficiency of RT-PCR amplification with degraded RNA extracted from long-term preserved unbuffered formalin-fixed, paraffin-embedded tissue specimens (for 19 to 21 years) is improved by the heating of RNA in citrate buffer prior to cDNA synthesis. This enhanced efficiency was possibly caused by RNA modification elimination and subsequent RNA template activity restoration.
Fragment sizes of ~60 bp can be amplified successfully at a rate of ~80% by RT-PCR, even when using RNA extracted from archival formalin-fixed, paraffin-embedded tissue samples stored for a long period. However, as in the case of amplification of the N-ras gene in two samples used in this study, archival tissue samples still remain in which a weak band is only vaguely observed or not detected even when fragment size by RT-PCR amplification is ~60 bp.
Furthermore, when amplicon size is very small, it is often difficult to design primers in restricted regions such as fusion points. Therefore, increased efficiency of RT-PCR amplification by the preheating of RNA is most effective when only limited quantities of archival formalin-fixed, paraffin-embedded tissue samples are available for study or when the designing of primers in restricted regions cannot be avoided.
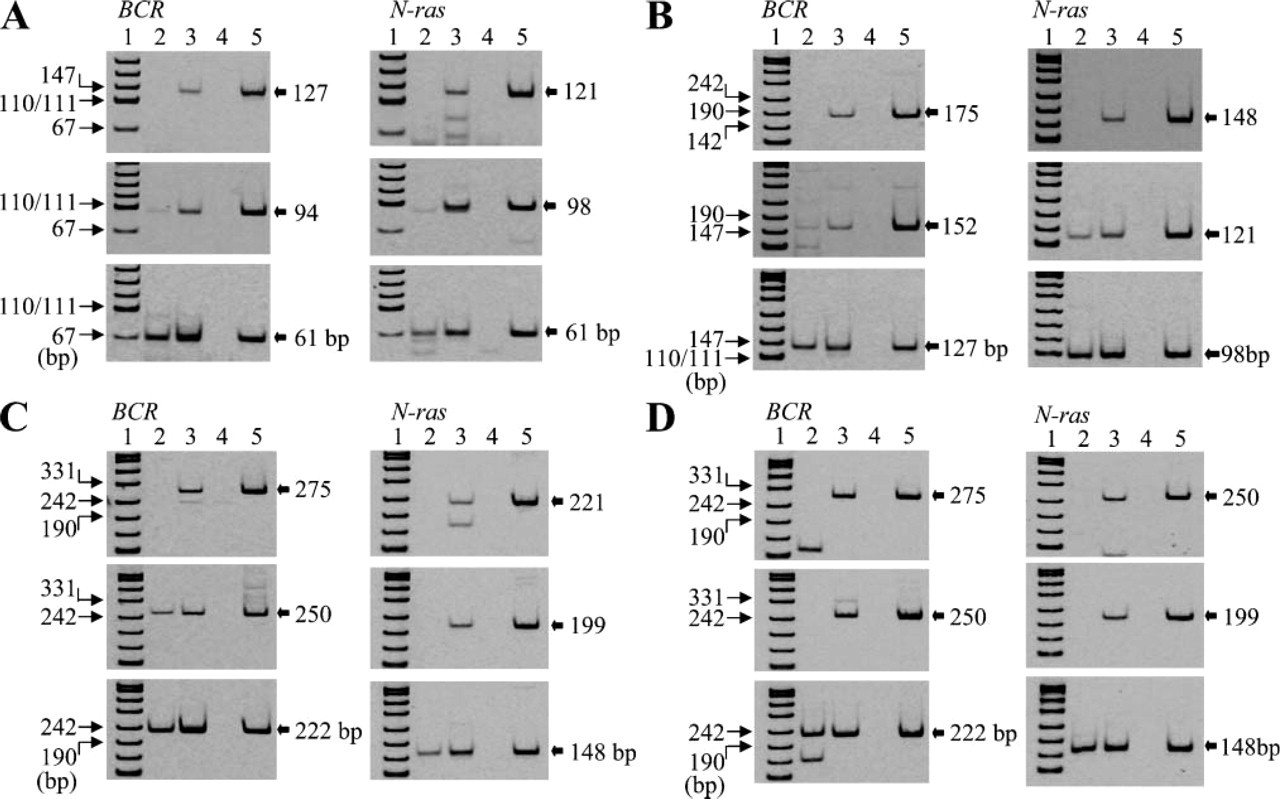
Application of preheating in citrate buffer (pH 4.0) to RT-PCR amplification of RNA extracted from the other four archival thyroid tissues (
Heat treatment with alkaline solution (pH 9-12) for DNA extraction from archival formalin-fixed, paraffin-embedded tissue increased the efficiency of DNA extraction, resulting in enhanced PCR amplification (Shi et al. 2002,2004). Our results demonstrate that heat treatment in citrate buffer with pH ranging from 3 to 6.5 improves to some extent the efficiency of RT-PCR amplification of RNA extracted from archival formalin-fixed, paraffin-embedded tissue, whereas treatment of RNA with pH solution ranging from 9 to 10 reduced the efficiency of RT-PCR amplification. These findings indicate that RNA or DNA modification induced by formalin may be removed more efficiently by preheating in acidic or alkaline buffer compared with neutralized buffer. The efficiency of RT-PCR amplification enhanced by RNA preheating in citrate buffer (pH 3-6.5) may be due to the fact that RNA is relatively stable in weak acidic solution but unstable in alkaline solution.
Treatment with highly concentrated citrate buffer reduced the efficiency of RT-PCR amplification of RNA compared with non-treated RNA. In our experiment, incubation time of 30-60 min in citrate buffer with pH 4.0 was the most efficient method for RT-PCR amplification. A longer incubation time, such as 2 hr, resulted in slightly decreased efficiency of RT-PCR amplification, suggesting that degradation of RNA may occur to some extent during the long preheating in citrate buffer at 70C.
Preheating of RNA in citrate buffer resulted in improved efficiency of RT-PCR amplification in all five archival tissue specimens examined, suggesting that this method is useful for molecular analyses of long-term preserved tissue specimens. This technique will enable the qualitative analysis such as DNA rearrangement using degraded RNA extracted from archival unbuffered formalin-fixed, paraffin-embedded tissue specimens that have been stored for more than several decades. It will shed light on the retrospective studies of rare cancers or cancers associated with exposure to uncommon past events such as Thorotrast treatment, nuclear power station accidents, or atomic bombings.
Footnotes
Acknowledgements
The Radiation Effects Research Foundation (RERF), Hiroshima and Nagasaki, Japan is a private, non-profit foundation funded by the Japanese Ministry of Health, Labor and Welfare (MHLW) and the U.S. Department of Energy (DOE), the latter through the National Academy of Sciences. This publication was supported by RERF Research Protocol (RP) No. 5-02 and in part by Grant-in-Aid for Japan-U.S. Cooperative Science Program Joint Project from the Japan Society for the Promotion of Science, for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and for Cancer Research from the Ministry of Health, Labor and Welfare of Japan.
We thank Shiho Yano and Kanya Hamasaki for excellent technical assistance.