
Abstract
Proteases are essential for protein catabolism, regulation of a wide range of biological processes, and in the pathogenesis of many diseases. Several techniques are available to localize activity of proteases in tissue sections or cell preparations. For localization of the activity of matrix metalloproteinases, in situ zymography was introduced some decades ago. The procedure is based on zymography using SDS polyacrylamide gels containing gelatin, casein, or fibrin as substrate. For in situ zymography, either a photographic emulsion containing gelatin or a fluorescence-labeled proteinaceous macromolecular substrate is brought into contact with a tissue section or cell preparation. After incubation, enzymatic activity is revealed as white spots in a dark background or as black spots in a fluorescent background. However, this approach does not allow precise localization of proteinase activity because of limited sensitivity. A major improvement in sensitivity was achieved with the introduction of dye-quenched (DQ-)gelatin, which is gelatin that is heavily labeled with FITC molecules so that its fluorescence is quenched. After cleavage of DQ-gelatin by gelatinolytic activity, fluorescent peptides are produced that are visible against a weakly fluorescent background. The incubation with DQ-gelatin can be combined with simultaneous immunohistochemical detection of a protein on the same section. To draw valid conclusions from the findings with in situ zymography, specific inhibitors need to be used and the technique has to be combined with immunohistochemistry and zymography. In that case, in situ zymography provides data that extend our understanding of the role of specific proteinases in various physiological and pathological conditions. (
A
Initially, proteases were considered as hydrolytic enzymes that were associated with protein catabolism, but it is now widely accepted that the highly specific hydrolysis of peptide bonds can regulate a wide range of biological processes (e.g., via processing of bioactive peptides) in all living organisms. This highly specific substrate cleavage is referred to as proteolytic processing, which regulates the activity and the compartmentalization of many proteins and therefore of many cellular processes (Barrett et al. 1998; Lopez-Otin and Overall 2002). Dysregulation of protease expression and in particular of their activity is involved in various pathological conditions, such as cardiovascular and neurodegenerative diseases, arthritic diseases, infection, and cancer. Therefore, proteases are attractive potential therapeutic targets.
Various techniques are used to determine the presence of proteases in tissues. Northern blotting analysis and RT-PCR are applied to quantify mRNAs in tissue extracts, whereas in situ hybridization (ISH) is used to localize mRNA in cell preparations or tissue sections. However, transcriptional activity does not necessarily reflect the amount and activity of the protein product of a certain gene. Western blots and immunohistochemistry (IHC) are used to determine the amount and localization of the protease protein but do not provide information about the activity of a protease because proteases are synthetized in an inactive proform or preproform that requires proteolytic processing for activation. Moreover, endogenous protease inhibitors can bind proteases and inhibit them. Biochemical techniques have been developed to detect protease activity in tissue extracts. However, homogenization of tissues for these assays does not allow localization of enzyme activity. In addition, extraction procedures can artifactually activate enzymes or cause interactions of active enzymes with their respective inhibitors when they are localized in different compartments in intact tissues. Therefore, techniques to localize specific proteolytic activity in cell preparations or tissue sections may provide crucial additional information on the exact role played by proteases in various physiological and pathological conditions.
Metabolic Mapping of Protease Activity
Over 30 years ago, Robert E. Smith and colleagues developed synthetic substrates for specific proteinases with the leaving group 4-methoxy-2-naphthylamine (MNA; Smith et al. 1972; Smith and Van Frank 1975). The substrates consist of the MNA-leaving group attached to an amino acid sequence specific for the proteinase under study (Lojda 1984). The latter can be coupled after proteolytic removal of the amino acids either to a diazonium salt, such as Fast Blue BB, to give a colored final reaction product (Lojda 1984), to hexazotized pararosanilin or new fuchsin for electron microscopic purposes (Schroeder and Gossrau 1982), or to 5-nitrosalicylaldehyde, which results in a yellow fluorescent reaction product (Dolbeare and Smith 1977). Figure 1 shows the ultrastructural localization of cathepsin B activity in lysosomes of rat liver parenchymal cells, as demonstrated with Z-ala-arg-arg-MNA as substrate and hexazotized pararosanilin as coupling reagent. The procedure was performed as described by Schellens et al. (2003).
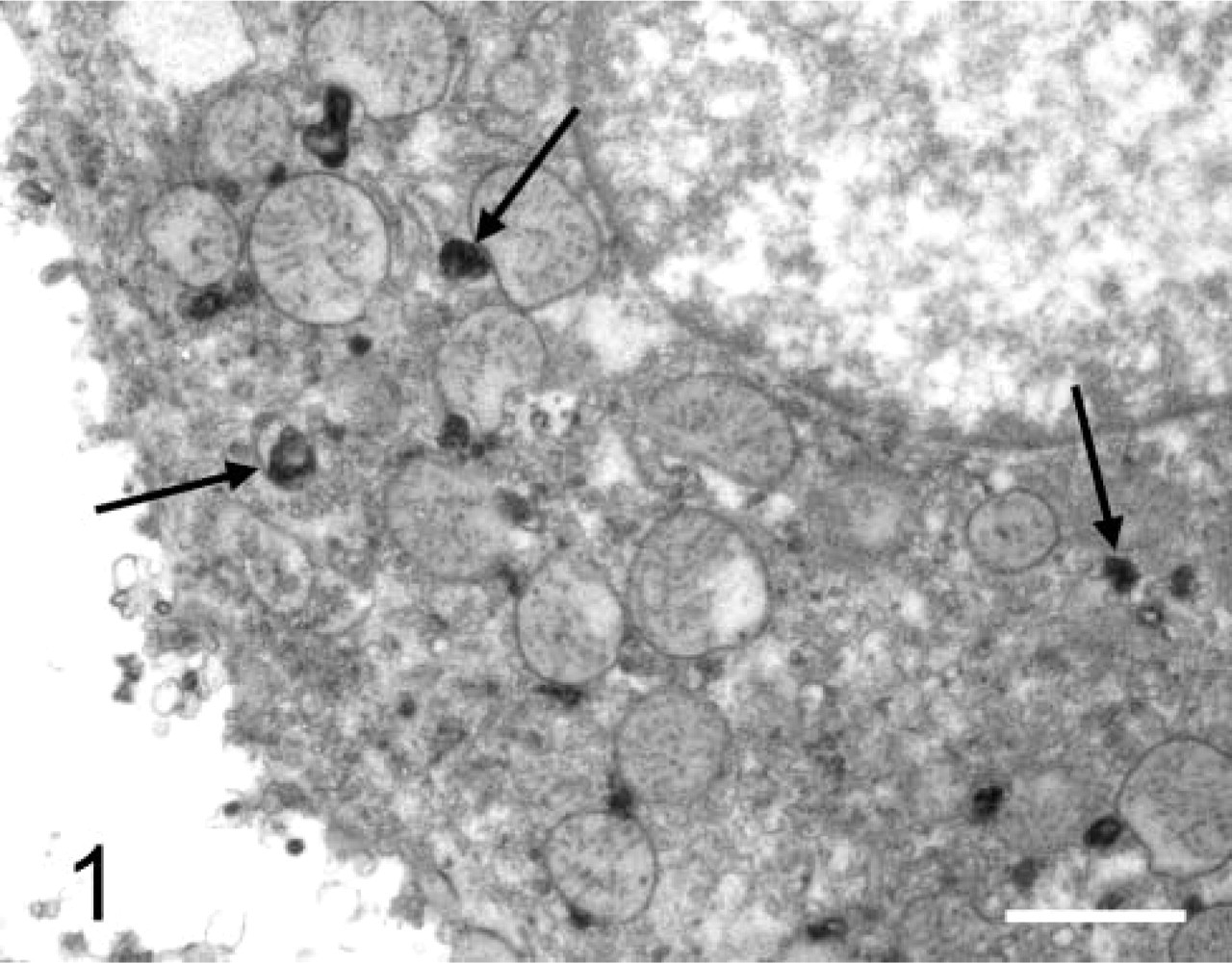
Electron micrograph of an unfixed, permeabilized, isolated liver parenchymal cell incubated for demonstration of cathepsin B activity with Z-ala-arg-arg-4-methoxy-2-naphthylamide as substrate and hexazotized p-rosanilin as coupling reagent. After incubation, cells were fixed in a mixture of glutaraldehyde and formaldehyde, postfixed in a solution of osmium tetroxide, and embedded in Epon. Final reaction product is localized exclusively in lysosomes. Bar = 0.5 μm.
Diazonium salts have been applied in simultaneous coupling methods for localization of various proteinases and peptidases (Lojda 1984). However, cysteine proteinases cannot be demonstrated very well with the simultaneous coupling method because these proteinases require SH groups for their activity and SH groups destroy diazonium salts. Moreover, diazonium salts strongly inhibit the activity of proteinases non-competitively. Therefore, either the fluorescence method with the use of 5-nitrosalicylaldehyde (Van Noorden et al. 1987) or a postcoupling method with Fast Blue BB as coupling reagent (Van Noorden et al. 1989) is the method of choice to localize activity of cysteine proteinases. The synthetic substrates for proteases with MNA as leaving group that are commercially available are listed in Table 1.
Yi et al. (2001) applied synthetic fluorogenic substrates with 7-amino-4-trifluoromethylcoumarin (AFC) as leaving group to demonstrate protease activities in the wound of swine skin, which approach was proposed by Lojda (1996). The substrate was incorporated into agarose so that the water-soluble fluorescent tag AFC could be properly localized. Substrates with AFC as leaving group are commercially available for many proteases. However, the activity of only a limited number of proteases has been localized thus far (Lojda 1996; Yi et al. 2001). It should be emphasized that small peptide substrates (with either MNA or AFC as leaving group) are not necessarily very specific. There is considerable overlapping in the affinity of different proteinases for various peptide substrates.
Synthetic substrates with 4-methoxy-2-naphthylamine as leaving group for metabolic mapping of proteases

It is becoming increasingly recognized that enzymes may behave differently in living cells and tissues than in frozen or fixed cells or tissue sections. Therefore, techniques are being developed for the detection of protease activities, such as those of dipeptidyl peptidase IV (DPPIV) and cathepsin B in living cells (Van Noorden et al. 1997,1998). A review on fluorogenic substrates available for metabolic mapping in living cells was recently published (Boonacker and Van Noorden 2001). Rhodamine-based fluorogenic dipeptide substrates were first synthesized by Leytus et al. (1983) for aminopeptidase, DPPIV, cathepsin B, cathepsin K, and cathepsin L. Recently, a new type of fluorogenic substrate for proteases has been synthetized based on the leaving group cresyl violet (Van Noorden et al. 1997,1998; Lee et al. 2003). The methods to localize protease activity in living cells using cresyl violet-based and rhodamine-based substrates have been critically evaluated by Boonacker et al. (2003).
Fluorogenic substrates for caspases are based on peptide sequences that are 18 amino acids long containing motifs that are specifically recognized by caspases and two identical fluorophores covalently attached near their termini. In such dimers, the fluorophore fluorescence is 90% quenched and fluorescence is generated when the substrate is cleaved (Komoriya et al. 2000; Kohler et al. 2002). The same type of substrate with quenched fluorogenic properties is also available for cathepsin D and MMP-2 (Bremer et al. 2001a, b). Thus far, these methods have been applied to living cells. Whether the localization of the final fluorescent reaction product is precisely enough to localize protease activity in tissue sections has yet to be established.
Proteolysis in tumor cells has been used by Weissleder et al. (1999) to develop an in vivo imaging system for tumors. An optimally quenched near-infrared fluorescence (NIRF) probe generates a strong NIRF signal after proteolytic activation. The probe is cleaved by lysosomal cysteine and serine proteases. This approach may also provide perspectives for localization of protease activities in tissue sections.
An entirely different approach to demonstrate activated caspases in living cells is the use of Caspa Tags, which are carboxyfluorescein-labeled fluoromethylketone inhibitors (Kohler et al. 2002). These cell-permeable inhibitors bind more or less specifically and irreversibly to the active site of caspases. Because the active site is available only in mature caspases, the Caspa Tags can exclusively stain cells containing the processed caspases of interest and the probes will not accumulate in normal (non-apoptotic) cells. This principle may have applications for the demonstration of other active proteases as well.
In conclusion, reliable techniques are available for the demonstration of activities of cysteine proteinases, serine proteinases, and aspartic proteinases using artificial substrates that contain small numbers of peptides, but demonstration of activities of MMPs with these small substrates had only limited success owing to the large numbers of peptide bonds that can be cleaved by MMPs and the overlap among different MMPs.
Zymography
Zymography is a simple, sensitive, quantifiable, and functional approach for the analysis of proteolytic activity in cell and tissue extracts, which was introduced more that 20 years ago (Heussen and Dowdle 1980). It is widely used to study extracellular matrix (ECM)-degrading enzymes, in particular the MMPs. MMPs are zinc-dependent endopeptidases capable of degrading ECMs, including the basement membrane. The MMP family consists of at least 26 members and has been classified into subgroups on the basis of substrate preference and molecular structure. These are interstitial collagenases, gelatinases, stromelysins, and matrilysins, although all enzymes have overlapping substrate specificity. A recently discovered MMP family consists of membrane-type MMPs that are characterized by a transmembrane domain or a GPI-anchor (Visse and Nagase 2003).
The standard method for zymography is based on the use of SDS-polyacrylamide gels co-polymerized with a protein substrate, in particular gelatin, casein, or fibrin. Proteases that have the ability to renature after removal of SDS and to exert proteolytic activity on a co-polymerized substrate can be analyzed with this method. MMP-2 (gelatinase A, 72 kD) and MMP-9 (gelatinase B, 92 kD) can be detected on gelatin zymograms and MMP-7 on casein gels. Coomassie Blue staining of the gel reveals sites of proteolysis as white bands on a dark blue background. Figure 2 shows a zymogram of a homogenate of a tumor of colon cancer cells in mouse liver containing four bands responsible for gelatin breakdown. Based on the molecular weights, these bands reflect inactive and active MMP-2 and inactive and active MMP-9 (Ackema, unpublished results). Polyacrylamide gel co-polymerization with plasminogen and gelatin allows detection of the plasminogen activators urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA) as plasmin generated by uPA and/or tPA degrades gelatin (Figure 3; Leber and Balkwill 1997). Casein zymography has been used as an alternative assay for fibrinolytic enzymes because of the ability of plasmin to degrade casein. Kim et al. (1998) introduced fibrin zymography to detect fibrinolytic enzymes by incorporating fibrinogen and thrombin into SDS polyacrylamide gels. Although gelatin and casein are satisfactory substrates for plasmin, all fibrinolytic enzymes are not able to cleave these substrates as plasmin is. Apparently, fibrin is an in vivo substrate for plasmin and plasmin-like enzymes that can be demonstrated reliably with fibrin zymography.
Zymography offers advantages over other methods, such as ELISA. Expensive materials are not required (e.g., antibodies) and proteases with different molecular weights showing activity towards the same substrate can be detected and quantified on a single gel. For example, MMPs are released from cells in a proteolytically inactive proform (zymogen) which is approximately 10 kD larger than the activated form. Because the proform becomes activated during the process of denaturation and renaturation after gel electrophoresis, the active form and the originally inactive forms degrade gelatin, and both forms can therefore be detected on zymograms. In addition, MMPs in solution are often associated with endogenous tissue inhibitors of metalloproteases (TIMPs). During electrophoresis the inhibitors dissociate from the MMP and do not interfere with detection of the enzymatic activity. On the other hand, sandwich ELISA can discriminate between MMP/TIMP complexes and free MMPs, resulting in determination of a potential active fraction (Zucker et al. 1992; Ratnikov et al. 2002; Catterall and Cawston 2003).
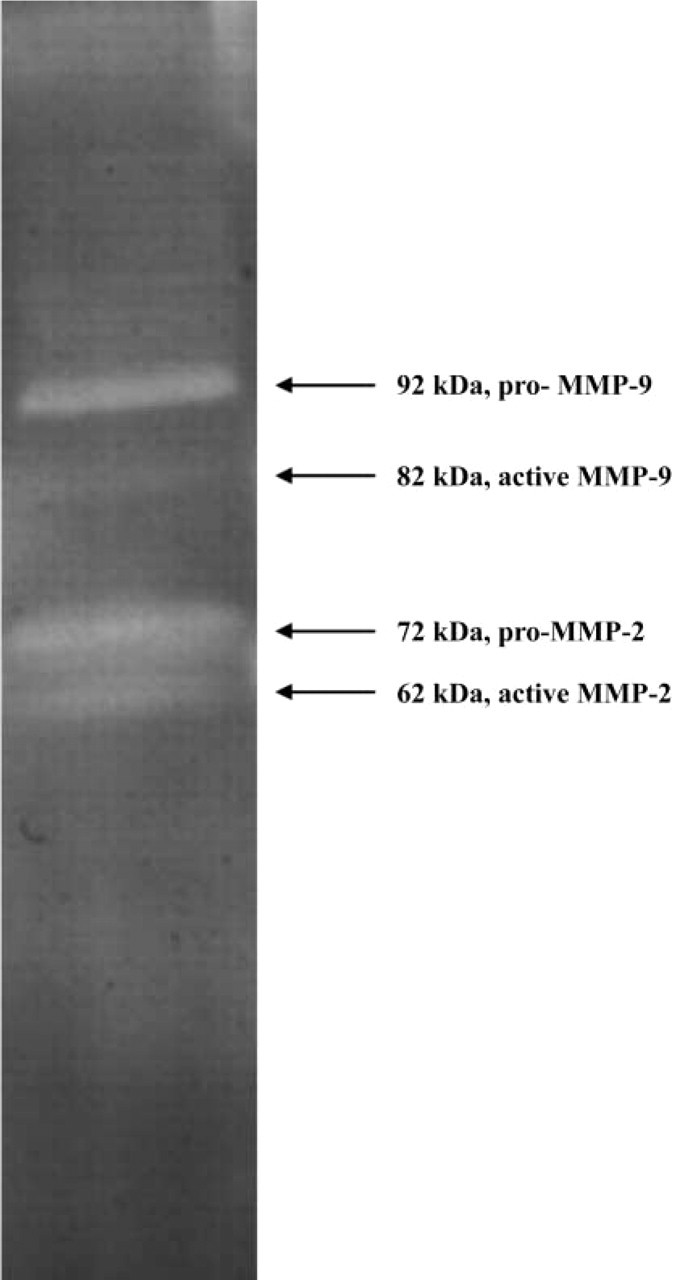
Gelatin zymography of a homogenate of colon cancer metastases in mouse liver. ProMMP-9 (92 kD), proMMP-2 (72 kD), active MMP-9 (82 kD), and active MMP-2 (62 kD) are present.
It can be concluded that zymography enables the detection of protease activity in cell or tissue homogenates using gelatin, casein, or fibrin as substrates. On the basis of molecular weight markers, the molecular weight of the proteolytic band can be determined, and by comparison with recombinant proteins and the use of specific protease inhibitors the type of protease can be established. However, information on the localization of the proteolytic activity in cells or tissues cannot be obtained on the basis of zymography.
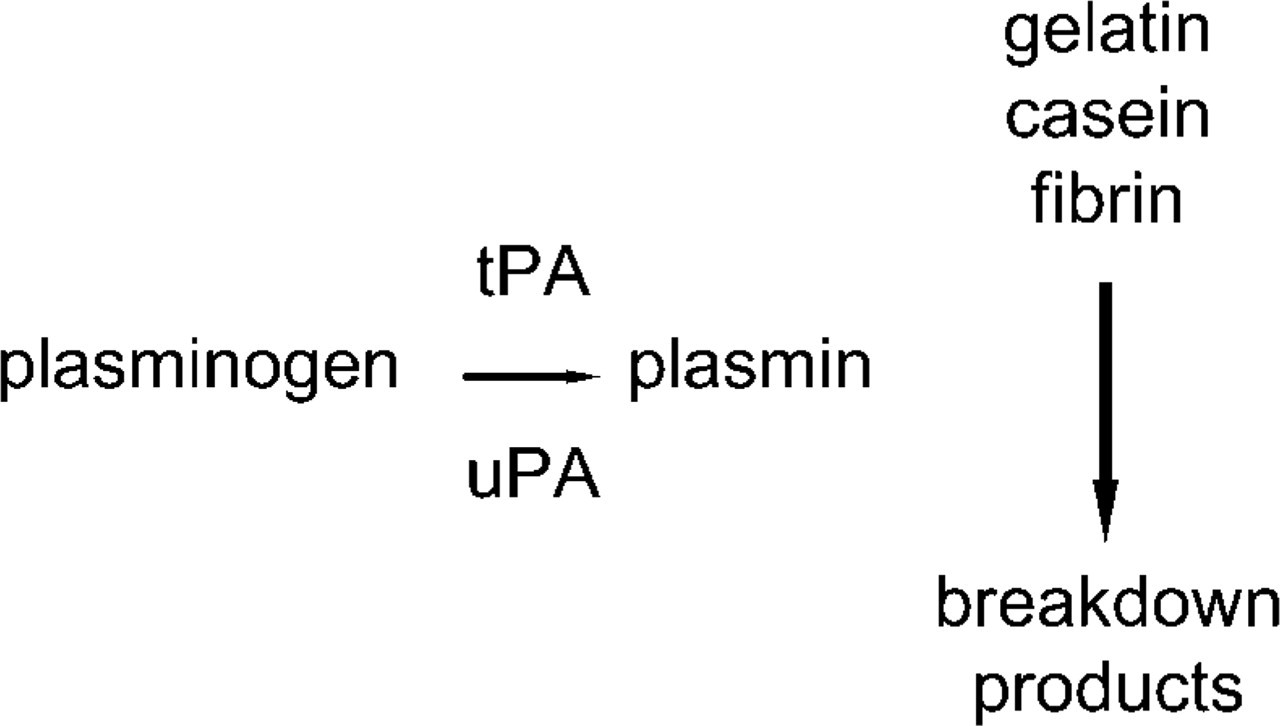
Principle of detection of activity of urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA).
In Situ Zymography of Gelatinase Activity
During the past decade, in situ zymography has been applied to localize gelatinase activity in tissue sections (Galis et al. 1994). This method is based on the principle of gel substrate zymography. Galis et al. (1994, 1995) used gelatin that contains fluorescein or an autoradiographic emulsion as substrate layer on cryostat sections of human atherosclerotic plaques to assess gelatinolytic activity in situ. Autoradiographic emulsions were introduced because gelatin is the main component of the emulsion and gelatinolytic activity during incubation results in decreased amounts of silver in specific sites that can be visualized by photographic development. Microscopy reveals transparent spots on top of the areas with gelatinolytic activity against a black background (Galis et al. 1994). Fluorescein-coupled gelatin was introduced to try to improve the precision of localization of gelatinolytic activity because disappearance of fluorescence indicates areas with gelatinolytic activity (Galis et al. 1995). Because fluorescein is a fluorophore that does not have optimal fluorescent properties for microscopy, Oregon Green conjugates were introduced, which have similar spectral characteristics as fluorescein, but their fluorescence is more photostable and less pH-dependent than fluorescein (Pirila et al. 2001; Faia et al. 2002).
The principle introduced by Galis et al. (1995) has been modified in the procedure to demonstrate breakdown of gelatin differently. Instead of fluorescent gelatin, some authors used pure gelatin that was stained after incubation with Ponceau S (Loy et al. 2002), amido black (Ikeda et al. 2000; Furuya et al. 2001), or Biebrich Scarlet (Wada et al. 2003). Another approach based on zymography was used by Kurschat et al. (2002), who incorporated gelatin into polyacrylamide gels of 50 μm thickness, which were brought into contact with unfixed cryostat sections. After incubation, sections and gels were separated and gels were stained with Coomassie Blue. All the approaches have in common the fact that a decrease in staining intensity is a reflection of gelatinolytic activity. Details of the principles of photographic emulsion-based and fluorescently-labeled substrate-based in situ zymography were recently reviewed by Yan and Blomme (2003). These authors concluded that the photographic emulsion technique was more sensitive than the fluorescent gelatin principle.
The approach of gelatin in situ zymography has been applied to study involvement of gelatinolytic activity in many (patho)physiological processes in tissues such as arteries (Knox et al. 1997; Bruno et al. 1998; Faia et al. 2002), veins (Fernandez et al. 1998; George et al. 1998; Kranzhofer et al. 1999), heart (Tyagi et al. 1996; Robert et al. 1997), lung (Pardo et al. 1996; Leco et al. 2001), skin (Fisher et al. 1997; Krejci-Papa and Paus 1998), eye (Zhou et al. 1998; Hanyu 1999), equine hoof (Mungall et al. 1998; Mungall and Pollitt 1999,2001), joints (Freemont et al. 1999; Yamanaka et al. 2000), colon (Tarlton et al. 2000), muscle (Kieseier et al. 2001), nerve (Duchossoy et al. 2001; Siebert et al. 2001), ovary (Curry et al. 2001; Khandoker et al. 2001), gingiva (Pirila et al. 2001), adipocytes (Maquoi et al. 2002), and endometrium (Zhang and Salamonsen 2002; Zheng et al. 2002). All studies indicate that gelatinases have a role in remodeling and/or degradation of the ECM.
Based on the high-level expression and proenzyme activation of gelatinases in tumors, in situ zymography has been used to study the involvement of gelatinolytic activity in cancer progression. Gelatinolytic activity was demonstrated in a series of human malignancies such as those of ovary (Furuya et al. 2001; Lengyel et al. 2001), cervix (Minami et al. 2003), breast (Iwata et al. 2001), lung (Ikeda et al. 2000; Kaji et al. 2003), thyroid (Nakamura et al. 1999), esophagus (Koyama et al. 2000), oral cavity (Shimada et al. 2000), brain (Nakada et al. 1999), kidney (Kamiya et al. 2003), and liver (Kaneyoshi et al. 2001). In almost all cases, MMP-2 appeared to be responsible for gelatinolytic activity and the activity was presumed to be related to the invasive and metastatic properties of the cancers.
The approach that was used in these studies was reduction in staining intensity of the gels on top of the sections. This approach has two major disadvantages. First, sensitivity of reduction in staining intensity is less than that of formation of staining and, second, it is doubtful whether this principle can be used for quantitative purposes (Thomas et al. 1998; Mungall and Pollitt 2001). Therefore, other procedures were developed in which a colored or fluorescent product was formed at the site of gelatinolytic activity. Ratnikov et al. (2000) introduced biotinylated gelatin as a substrate to demonstrate gelatinolytic activity in solution. After cleavage of the substrate, the proteolytic fragments bearing the biotin moieties are captured by streptavidin coated on the plastic surface of a 96-well microtiter plate. Activity of horseradish peroxidase conjugated to streptavidin is measured. This biotinylated gelatin may also have perspectives for in situ localization when diaminobenzidine or aminoethylcarbazole is used to localize peroxidase activity.
The application of gelatin as substrate for in situ zymography has the advantage that, as far as we know, only MMP-2 and MMP-9 have a high affinity for this substrate. However, definite conclusions about the specific enzyme(s) responsible for gelatin breakdown can be drawn only when selective inhibitors are used and the in situ zymography is combined with gelatin zymography and IHC of MMP-2, MMP-9, and other potential gelatin-degrading enzymes. Gelatin zymography enables the assessment of molecular weights of the proteins that degrade gelatin. Moreover, differences in molecular weight of proenzymes and activated enzymes allow estimation of relative amounts of proenzymes and active enzymes in homogenates of tissues under study.
It can be concluded that in situ zymography with gelatin as substrate enables the localization of MMP activities, but thus far precise localization is not possible.
In Situ Zymography with Quenched Fluorogenic DQ-gelatin
Precise localization of gelatinase activity in sections and cells became possible with the introduction of dye-quenched (DQ)-gelatin, which is gelatin that is heavily labeled with FITC molecules so that its fluorescence is quenched (Oh et al. 1999; Curry et al. 2001; Duchossoy et al. 2001; Goodall et al. 2001; Lindsey et al. 2001; Teesalu et al. 2001; Wang and Lakatta 2002; Zhang and Salamonsen 2002; Mook et al. 2003; Platt et al. 2003; Lee et al. 2004). After cleavage of DQ-gelatin by gelatinolytic activity, fluorescent peptides are produced that can be visualized against a weakly fluorescent background (EnzCheck; Molecular Probes, Eugene, OR). The substrate was developed for assaying protease activity in solutions, but the substrate has properties that enable localization of protease activity in cells or tissues under certain conditions. Oh et al. (1999) incubated unfixed cryostat sections of developing optic nerves and cultured live oligodendrocytes with an aqueous medium containing DQ-gelatin. After overnight incubation without any further fixation or washes, gelatinolytic activity was localized and photographed. This procedure enabled localization of MMP-2 and MMP-9 activity in developing optic nerves. It appeared that the activity had a similar distribution pattern as myelin basic protein. Activity was also found pericellularly at growing tips of oligodendrocytes. Formation of fluorescent peptides was prevented by addition of phenanthroline or TIMP-1 to the incubation media, which indicates that MMP activity was demonstrated. Parallel investigations on MMP-2 and MMP-9 of oligodendrocytes with zymography led to the conclusion that MMP-9 was the gelatinase involved in myelin formation by oligodendrocytes. The same procedure was recently applied by Lee et al. (2004), who localized gelatinolytic activity due to MMP-9 in pyramidal and granular neurons of the hippocampus after ischemia. An important role for MMP-9 in the pathogenesis of neuronal damage was proposed. Curry et al. (2001) added 1% agarose to DQ-gelatin in analogy with the principle introduced by Galis et al. (1995). After spreading this solution on glass slides and gelling at 4C, unfixed cryostat sections were mounted on the gelatin substrate, coverslipped, and incubated for 20 hr at 37C. Fluorescence was detected in rat ovaries during follicular growth, ovulation, and early luteal formation. Production of fluorescence was prevented by EDTA and ilomastat, a synthetic MMP inhibitor. Therefore, it was concluded that gelatinolytic activity by MMPs was demonstrated in these tissues. Teesalu et al. (2001) used DQ-gelatin, agar, and unfixed cryostat sections to localize gelatinolytic activity in inflammatory lesions caused by experimental autoimmune encephalomyelitis and demonstrated that MMP-9 was responsible for the in situ gelatin breakdown on the basis of parallel zymography experiments.
MMP-9 activity was detected in the vicinity of infiltrating neutrophils in canine myocardium subjected to ischemia and reperfusion with the use of DQ-gelatin applied to unfixed cryostat sections (Lindsey et al. 2001). Gelatinolytic activity was inhibited by both EDTA and neutralizing MMP-9 antibody. Moreover, gelatinolytic activity was not observed in non-ischemic myocardium. It was concluded that infiltrating neutrophils were the source of active MMP-9.
MMP-2 activity was localized on elastin fibers in blood vessel walls after incubation of cryostat sections of inferior mesenteric veins of patients with abdominal aortic aneurysms with DQ-gelatin (Goodall et al. 2001). Phenanthroline inhibited production of fluorescence and gelatin zymography demonstrated the presence of active MMP-2. The authors concluded that MMP-2 plays a primary role in aneurysm formation. However, it should be noted that autofluorescence of elastin fibers may interfere with detection of fluorescence due to gelatinolytic activity.
Duchossoy et al. (2001) combined in situ zymography using DQ-gelatin with immunodetection of laminin to study the role of MMPs in spinal cord injury. For this purpose, unfixed spinal cord cryostat sections were incubated overnight in medium containing DQ-gelatin and antibody against laminin. After a rapid wash, sections were incubated with secondary antibodies and studied with fluorescence microscopy. Gelatinolytic activity was co-localized with laminin surrounding blood vessels in injured spinal cord and was detected in lesion sites in and around infiltrating cells. Preincubation with blocking antibodies against MMP-2 and MMP-9 strongly reduced fluorescence intensity, suggesting the involvement of gelatinase activity. The authors concluded that gelatinases play a role in blood–spinal barrier disruption, leukocyte infiltration, disruption of the ECM, and clearance of debris after spinal cord injury. Because it is difficult to imagine that soluble fluorescent peptides produced by cleavage of DQ-gelatin can be detected after a wash of the sections and additional incubation with secondary antibodies, it must be concluded that extracellular localization of gelatinase activity must be the result of binding of fluorescent peptides to MMPs in the ECM.
Zhang and Salamonsen (2002) were not successful in combining in situ zymography using DQ-gelatin and immunostaining of MMPs on the same section of human endometrium. This may be due to the fact that cryostat sections were fixed in formalin and 2% gelatin was added to the DQ-gelatin-containing incubation medium. It has been established that fixation with formaldehyde and therefore no doubt with formalin, completely inhibits gelatinolytic activity (Mook et al. 2003). Moreover, the addition of 2% gelatin to 0.01% DQ-gelatin may reduce the efficacy of breakdown of DQ-gelatin. In situ zymography without simultaneous IHC detection of MMP-2 and MMP-9 resulted in findings on the basis of which it was concluded that MMPs were more active in menstrual endometrium compared with other stages of the cycle, which suggests that MMPs play a role in matrix degradation during menstruation. Wang and Lakatta (2002) demonstrated gelatinolytic activity to be increased in the wall of rat aortas in relation with aging using DQ-gelatin. However, detailed descriptions of the method they applied were lacking. It was concluded that activity mainly resulted from MMP-2 because activity was almost completely inhibited by a blocking antibody against MMP-2. The role of MMP-2 and MMP-9 in rat sciatic nerves after crush and during regeneration was established with in situ zymography with DQ-gelatin and gel zymography (Platt et al. 2003). The fluorescence due to gelatinase activity at the lesion site was prevented by addition of phenanthroline.
The first application to cancer of in situ zymography with DQ-gelatin was performed by Mook et al. (2003). The procedure was based on the incubation of unfixed cryostat sections of rat liver containing colon cancer metastases using a low-gelling temperature (LGT) agar- and DQ-gelatin-containing incubation medium. The time of incubation was only 60 min, the method could be combined with IHC on the same section, and the method was extensively tested for its specificity. Gelatinolytic activity due to MMP-2 was detected in stroma of colon cancer metastases (Figure 4).
It can be concluded that the use of DQ-gelatin instead of labeled or unlabeled gelatin is superior for in situ zymography because fluorescence is produced at sites of gelatinolytic activity instead of decreased staining intensity at gelatinolytic areas. However, the limitations described above for in situ gelatin zymography apply for DQ-gelatin as well. Moreover, autofluorescence of the tissue should be carefully inspected by incubation of tissue sections with incubation media that lack the substrate.
In Situ Zymography with Other Quenched Fluorogenic Substrates
Most studies performed thus far with in situ zymography using quenched fluorogenic substrates dealt with gelatinolytic activity. However, other quenched fluorogenic substrates are also available. These are DQ-collagen type I, DQ-collagen type IV, DQ-elastin, DQ-bovine serum albumin (BSA), DQ-ovalbumin, and DQ-casein (Jones et al. 1997). These substrates were initially developed to demonstrate activity of proteinases in solution. However, the substrates can in principle be used also to detect proteinase activity in unfixed cryostat sections or cell preparations when LGT-agar or another gelling compound is added to the incubation medium. It must be emphasized that development of other natural substrates of MMPs with quenched fluorescence, such as laminins and fibronectins, should be encouraged.
DQ-collagen type I was applied to fixed cryostat sections of endometrial biopsy specimens in the presence of gelatin (Zhang and Salamonsen 2002). Collagenase activity was observed in small foci within the tissue in all phases of the menstrual cycle, but collagenase activity was most abundant premenstrually and during the menstrual phase. Collagenase activity was localized extracellularly and was abolished when sections were pretreated with phenanthroline. It was concluded that MMP-1 was responsible for the breakdown of collagen type I because the protein showed a similar localization pattern. MMPs appear to play a critical role in matrix degradation during menstruation.
DQ-collagen type IV has been mainly applied to cells cultured on matrices containing the substrate (Horino et al. 2001; Sameni et al. 2001; Elner 2002; Premzl et al. 2003). Invasion of fibrosarcoma cells in gelatin matrix was accompanied by collagenolytic activity due to MMP activity (Horino et al. 2001). Sameni et al. (2000, 2001) investigated the site of proteolysis in a three-dimensional gelatin matrix embedded with DQ-collagen type IV and human cancer cells cultured on top of it. Cells at all levels in the matrix accumulated fluorescent degradation products of DQ-collagen IV intracellularly in vesicles. Based on findings using inhibitors, it was concluded that lysosomal proteinases, such as cathepsin B, are responsible for intracellular degradation of collagen type IV in human breast cancer cells, glioma cells, and colon cancer cells, and are involved in cancer cell invasion.
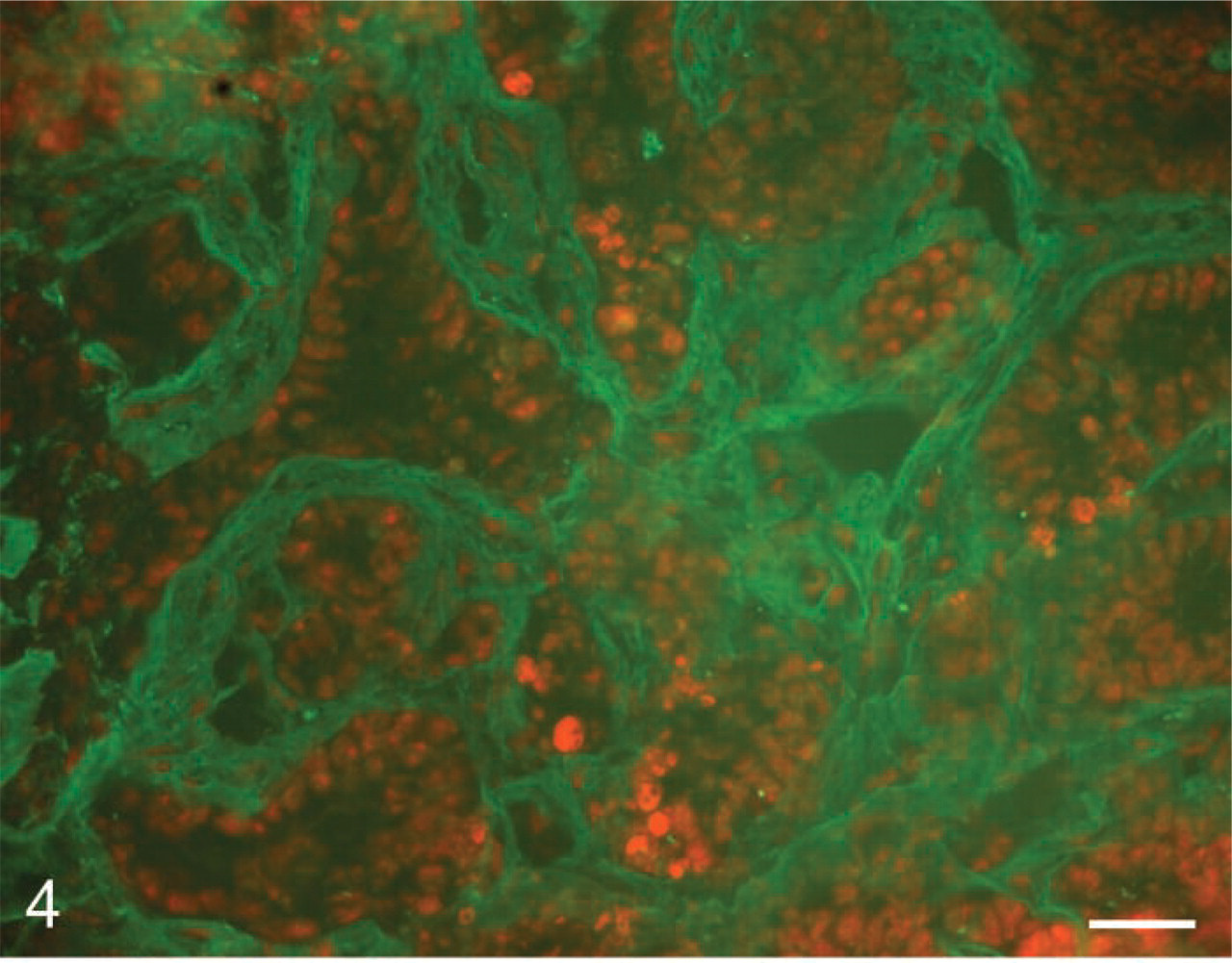
In situ zymography of gelatinolytic activity with DQ-gelatin as substrate in colon cancer metastasis in rat liver. Fluorescence due to gelatinolytic activity (green) was found in the extracellular matrix of tumors. Nuclei are shown in red. Bar = 35 μm.
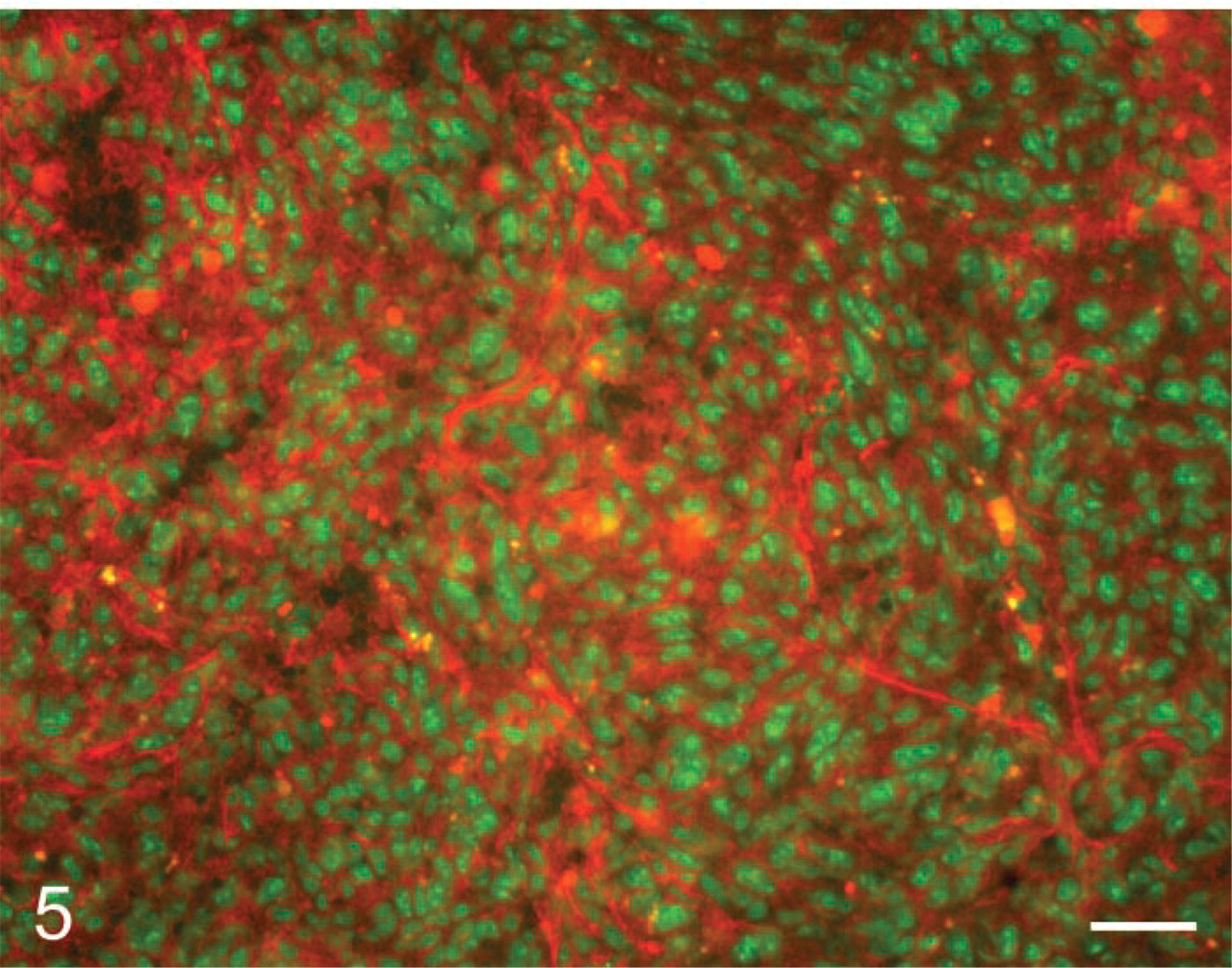
In situ zymography of urokinase-type plasminogen activator activity with Bodipy-casein as substrate and added plasminogen in colon cancer metastasis in mouse liver. Fluorescence due to caseinolytic activity (red) was found in intratumoral stroma of tumors. Nuclei are shown in green. Bar = 35 μm.
A similar approach was used by Premzl et al. (2003), who localized the breakdown of DQ-collagen IV by ras-transformed breast cancer cells during invasion in matrigel. Intracellular and extracellular cleavage of collagen type IV was detected that was largely due to intracellular and extracellular cathepsin B activities. Yan and Blomme (2003) demonstrated breakdown of DQ-collagen type IV in cryostat sections of rat tibia. Fluorescence was observed in the distal portion of the hypertrophic zone of growth plates of the tibia. MMP-9 was held responsible for collagenolytic activity because EDTA prevented the production of fluorescence, and protein and mRNA of MMP-9 were similarly localized as the fluorescence due to collagen type IV breakdown.
In situ zymography of uPA was introduced by Sappino et al. (1991) using plasminogen and casein as substrate. Plasminogen was converted by uPA to plasmin, which degraded casein. de Vries et al. (1995) demonstrated with this approach uPA activity in sprouting capillaries in melanomas. The production of fluorescent peptides from DQ-casein was recently used for the precise localization of uPA activity in unfixed cryostat sections of mouse liver with colon cancer metastases (Ackema, unpublished results). Figure 5 shows the localization of fluorescent breakdown products of Bodipy-casein in colon cancer metastases of mouse liver. DQ-BSA has also been used instead of DQ-casein as substrate for the localization of uPA activity in matrices containing fibrosarcoma cells (Horino et al. 2001) and retinal pigment cells (Elner 2002). The specificity of extracellular proteolysis of BSA by uPA was assessed by inhibition with PAI-1. BSA can be cleaved by many proteinases and specificity tests are therefore very important. Koblinski et al. (2000) showed that intracellular cathepsin B is responsible for the intracellular accummulation of degraded DQ-BSA in transfected fibroblasts grown on a gelatin matrix containing DQ-BSA.
Summarizing, dye-quenched fluorogenic natural substrates other than gelatin have mainly been used in gel matrices containing cultured cells and have rarely been applied to cryostat sections. The use of these substrates has potential for the localization of activity of proteinases, but it should be emphasized that proper controls must be performed to establish the proteinases involved, such as the use of inhibitors and combination with detection methods such as IHC and zymography.
In conclusion, precise localization of proteinase activity using natural substrates containing quenched fluorescence is a valuable tool to study its role in (patho)physiological processes. The value of the method may even increase when fluorescence production can be measured locally. Fluorescence should then be measured locally during incubation, and measurements should fulfill a series of criteria as formulated by Stoward (1980), as has been established for many other enzyme histochemical procedures (Van Noorden and Frederiks 1992). Moreover, in situ zymography localization should be combined with IHC, preferably on the same sections or at least on serial sections. Finally, zymography of cell or tissue homogenates should also be performed to assess the nature of the protein and, as a consequence, the type of proteinase that cleaves the substrate.
Protocol 1
This is the method of choice to detect gelatinolytic activity with in situ zymography and DQ-gelatin in cells or tissues.
Dissolve 1 g LGT agarose in 100 ml PBS, pH 7.45, under continuous stirring and heating in a water bath (80C) until a clear solution is obtained.
Store the clear agarose-containing solution at 4C in air-tight vials.
Heat the agarose-containing solution to 60C to obtain a clear solution before incubation.
Cool the desired volume of the solution to 37C before incubation.
DQ-gelatin (Molecular Probes) is dissolved in a concentration of 1 mg/ml in distilled water.
The DQ-gelatin solution is diluted 1:10 in the agarose-containing solution.
Use unfixed cells or unfixed cryostat sections of the tissue to be investigated (8–10 μm thick).
The DQ-gelatin agarose mixture (40 μl) is put on top of the dried cells or sections and covered with a coverslip of 24 × 40 mm.
The agar is gelled at 4C.
Incubation is performed for 1–24 hr at RT dependent on the enzyme activity.
Fluorescence of FITC is detected with excitation at 460–500 nm and emission at 512–542 nm.
Control incubations should be carried out on cells or serial cryostat sections by adding 20 mM EDTA or a selective MMP inhibitor to the incubation medium.
Cells or sections should be preincubated for 1 hr at RT with the different inhibitors dissolved in PBS.
The presence of autofluorescence in cells or sections should be tested by incubating in the agarose-containing medium that lacks DQ-gelatin.
Nuclei can be counterstained by adding DAPI (1 μg/ml) or propidium iodide (0.5 μg/ml) to the incubation medium.
Analyze the cells of sections: compare the fluorescence of FITC formed after incubation in the presence of DQ-gelatin with the fluorescence produced after incubation in the absence of DQ-gelatin or in the presence of DQ-gelatin and MMP inhibitors.
Protocol 2
This is the procedure of choice to detect gelatinolytic activity with in situ zymography in cells or tissues in combination with IHC.
Cells or cryostat sections are air-dried for 1 hr at RT.
Cells or cryostat sections are fixed in acetone for 10 min at RT. Crosslinking fixatives should be avoided, because they affect enzyme activity.
The IHC procedure for the desired antigen is performed according to standard procedures using a fluorescently labeled secondary antibody with spectral properties other than FITC.
The procedure as described in protocol 1 is performed to detect gelatinolytic activity.
Analyze the cells or sections: compare the localization of the fluorescence of FITC with that of the other fluorophore.
Footnotes
Acknowledgements
We are grateful to Prof Dr C.J.F. Van Noorden for most valuable comments on the manuscript. We wish to thank Ms H. Vreeling and Ms E. Ackema for providing their micrographs, Mr J. Peeterse for preparation of the micrographs, and Ms T.M.S. Pierik for careful preparation of the manuscript.