
Abstract
Gap junctions formed by connexins mediate cell–cell communication by electrical and chemical coupling. Recently, it has been shown that alterations in the phosphorylation state of the connexins result in functional alteration of cell–cell communication through gap junctions. Therefore, we focused on the association of alterations of phosphorylation state of connexin 43 (Cx43) with cardiac function in vivo. Rat hearts were transferred to Langendorff apparatus and submitted to hypoxia and then reoxygenated. In the control heart, Cx43 was phosphorylated and located at the intercalated disk. When the hearts were subjected to hypoxia, Cx43 at gap junctions was dephosphorylated and changed its localization to the entire plasma membrane. The area of cardiomyocytes stained with anti-phosphorylated Cx43 antibody was decreased in a time-dependent manner. Immunoblot data supported the decrease of phosphorylated Cx43 during hypoxia. ZO-1 did not change its localization at the intercalated disk during the hypoxic period. We also found that the area occupied by dephosphorylated Cx43 was correlated with the decrease of percent of rate-pressure product. These data indicate that dephosphorylation and redistribution of Cx43 is an early sign of cardiac injury after hypoxia. Detection of dephosphorylated Cx43 may serve as a diagnostic tool for examining ischemic heart disease.
T
Phosphorylation of gap junctional proteins appears to regulate channel function and the rates of channel assembly and turnover (Laird et al. 1991; Musil and Goodenough 1991; Oelze et al. 1995; Beardslee et al. 1998; Saffitz et al. 2000; TenBroek et al. 2001; Leykauf et al. 2003). Channel assembly in cardiomyocytes is also regulated by the interaction of Cx43 with other intercellular junction molecules including ZO-1 and cadherins (Tepass et al. 2000; Toyofuku et al. 2001; Barker et al. 2002; Ferreira-Cornwell et al. 2002; Luo and Radice 2003; Duffy et al. 2004). Phosphorylation of Cx43 has been described mainly on serine but also at tyrosine residues (Lampe and Lau 2000; Toyofuku et al. 2001; Thomas et al. 2003). Several protein kinases are able to influence junctional permeability. In vitro studies have revealed that protein kinase C or mitogen-activating protein kinase, which correlates with enhanced phosphorylation of Cx43 at serine residues, abolished gap junctional coupling, whereas c-Src, which phsophorylates tyrosine residues, inhibits junctional communication (Crow et al. 1990; Lampe et al. 2000; Giepmans et al. 2001; Toyofuku et al. 2001; Leykauf et al. 2003; Bao et al. 2004; Doble et al. 2004).
There has been speculation that gap junction remodeling contributes to arrhythmogenesis in diseased myocardium (Smith et al. 1991; Peters et al. 1997). It is known that ischemia induces dephosphorylation of Cx43 and translocation of the protein from the intercalated disk to the cytoplasm in myocytes (Beardslee et al. 2000). The remodeling of gap junctions in diseased heart might be accompanied by the change of phosphorylation state of Cx43 by several kinases or phosphatases (Ya et al. 1998; Lampe and Lau 2000; Gutstein et al. 2001; Leithe et al. 2003; Leykauf et al. 2003). However, the relationship between alterations in phosphorylation state of Cx43 and cardiac function in vivo remains unclear.
In this work we evaluated the alterations in phosphorylation state of Cx43 after hypoxic injury and its relation to cardiac function using an ex vivo perfusion system of rat heart.
Materials and Methods
Preparation and Perfusion Procedure
All procedures performed on laboratory animals were approved by the Institutional Animal Care Committee of Juntendo University School of Medicine. All animal experiments were carried out in compliance with the guidelines for animal experimentation of Juntendo University School of Medicine.
Male Wistar rats, weighing 280–320 g, were used. Their hearts were excised quickly, established on retrograde perfusion via the aortic cannula, and then perfused with modified Krebs–Henseleit solution (pO2 > 400 mmHg, 38C) as previously described (Okada et al. 2000). During hypoxic perfusion (pO2 < 50 mmHg), glucose was replaced by equimolar sucrose. Perfusion was done at a constant flow rate (13 ml/min) without recirculation. After a latex balloon was inserted through the mitral annulus into the left ventricular cavity, we monitored left ventricular heart rate (HR) and developing pressure (DP). Coronary effluent was collected for the quantification of released glutamic oxaloacetic transaminase (GOT).
Experimental Protocols
In this study we performed three protocols (Figure 1). The first examined hypoxia alone, the second was for hypoxia followed by 5-min reoxygenation, and the third for hypoxia followed by 30-min reoxygenation. After a 20-min stabilizing period, hearts received the following treatments: hearts were submitted to hypoxia for 5, 20, or 40 min (n=8 each). For the hypoxia plus reoxygenation group, hypoxia was followed by 5 (n=19) or 30 min (n=22) of reoxygenation. Control hearts (n=8) were submitted to 30-min normoxia following the stabilizing period.
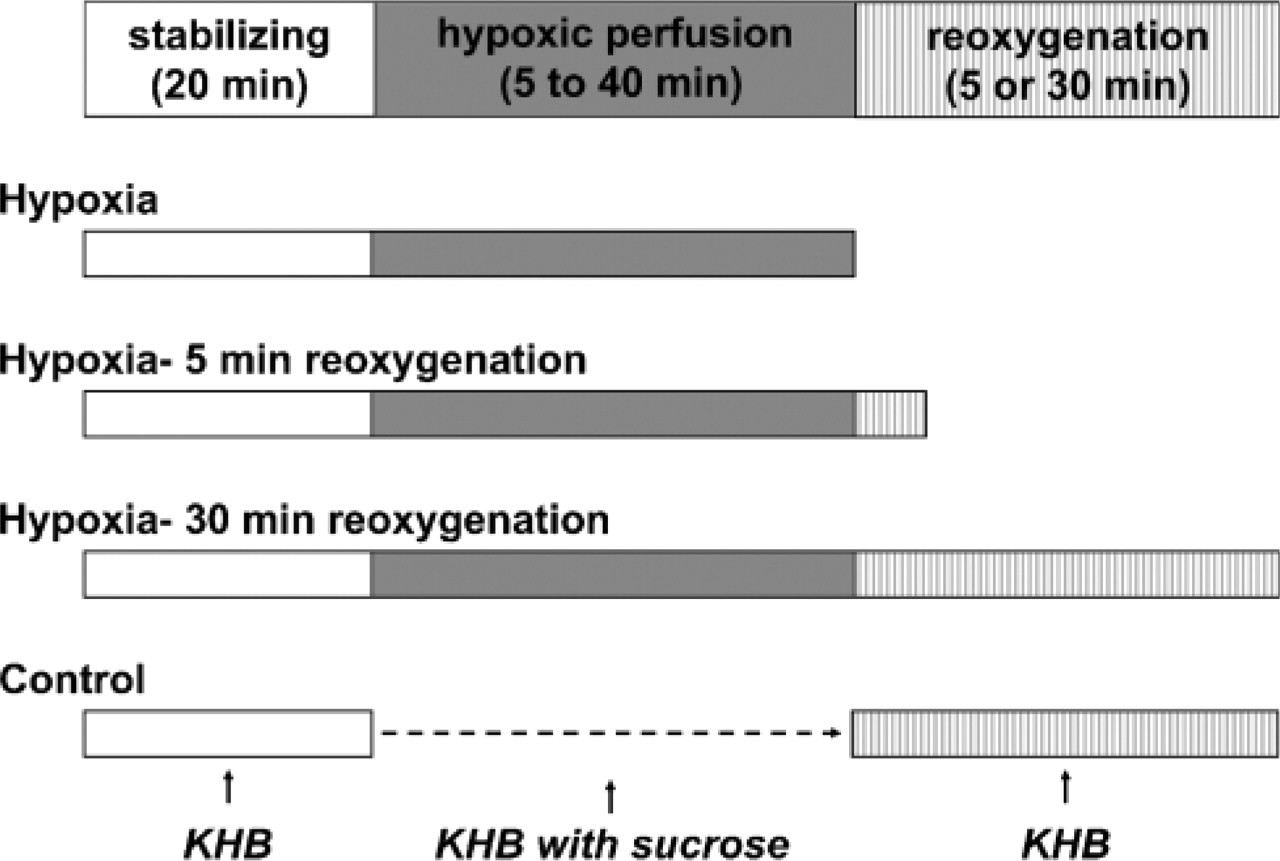
Experimental protocols. To examine the changes of phosphorylation status of connexin 43 (Cx43) during hypoxia and hypoxia–reperfusion injuries, we performed three different protocols: hypoxia, hypoxia–5-min reperfusion, and hypoxia–30-min reperfusion. In hypoxia group, hearts were exposed to hypoxia for 5, 20, and 40 min. In hypoxia–reoxygenation groups, hypoxic perfusion was followed by 5 or 30 min of reoxygenation. Control hearts were perfused with oxygenated solution for 30 min after the stabilizing period. KHB, Krebs-Henseleit buffer.
Immunofluorescence
At the end of perfusion experiments, hearts were slowly fixed with 4% paraformaldehyde (PFA) buffered with 0.1 M phosphate buffer (pH 7.4) injected through the aortic cannula under anesthesia with Nembutal. The left ventricle was then trimmed at the same level to include papillary muscle, and the cardiac pieces were immersed in PFA for 30 min. After washing with phosphate-buffered saline (PBS, pH 7.4), tissue pieces were dehydrated in PBS containing 10%, 15%, and 20% sucrose solution, respectively. These sections were embedded in optimal cutting temperature compound and prepared for 3-μm thickness with a cryostat (Jung Frigocut 2800E; Leica, Wetzlar, Germany). Sections were incubated with primary antibodies for 2 hr followed by secondary antibodies for 1 hr at room temperature. We used three kinds of antibodies that recognized differential phosphorylation status of Cx43 and one anti-ZO-1 antibody. Details of primary antibodies are listed in Table 1. TRITC- or FITC-conjugated donkey anti-mouse IgG or donkey anti-rabbit IgG was used as the secondary antibody (Jackson ImmunoResearch Laboratories; West Grove, PA).
Western Blot Analysis
Hearts were solubilized in PBS containing protease inhibitors (1 mM each antipain, benzamidine, leupeptin, pepstatin A, and PMSF), 1% SDS, and 5 mM EDTA, electrophoresed on 12% polyacrylamide gels, and transferred to nitrocellulose membranes. Blots were incubated with primary antibodies and then with horseradish peroxidase-conjugated secondary antibodies (BioRad; Richmond, CA) and detected using the ECL Western Blotting Detection System (Amersham; Arlington Heights, IL). Quantification of protein bands was done by densitometry with the use of QuantiScan software (Biosoft; Cambridge, UK).
List of primary antibodies

ap, phosphorylated form of Cx43.
bnp, non-phosphorylated form of Cx43.
Image Analysis
Fluorescence specimens were observed with a Leica DMR microscope. Images were captured with a Hamamatsu Orca-ER CCD camera and analyzed using AquaCosmos software (Hamamatsu Photonics; Hamamatsu, Japan).
Statistical Analysis
Statistical analysis was performed with the aid of commercially available software (Statcel 2 for Microsoft Excel; O.M.S., Saitama, Japan). Comparison of area analysis among groups was performed by Scheffe's F test, correlation between rate-pressure product (%RPP, HR X DP) and area analysis was performed by simple regression analysis.
Results
Physiological Assessment of Cardiac Function
To determine the functional change of isolated heart during hypoxia–reoxygenation, we recorded HR and DP, then calculated RPP: (HR X DP). GOT was measured during perfusion experiments. These parameters for each group during perfusion were expressed as the change of percentage from the baseline. The changes of %HR, %DP, %RPP, and GOT in the hypoxia–30-min reoxygenation group were shown as mean value in Figure 2. All materials were stabilized for 20 min. At the end of the stabilizing period, values of hemodynamic parameters were similar in all groups. In addition, all groups (hypoxia, hypoxia–5-min reoxygenation, and hypoxia–30-min reoxygenation) showed a similar tendency in these parameters during experiments.
During hypoxic perfusion, %HR, %DP, and %RPP were gradually decreased compared with control (CL). After reoxygenation, these parameters were increased and reached a plateau at the end of reoxygenation for 30 min and later did not change. Reoxygenation for > 30 min did not affect the recovery of these parameters examined in this study. %HR recovered to the prehypoxic level in all groups; thus, hypoxic perfusion time was varied (CL: 106.1 ± 5.2%, 5 min; 103.4 ± 2.3%, 20 min; 89.2 ± 5.6%, 40 min; 97.1 ± 6.9%, mean ± SE, respectively). There was no statistical significance among the groups.
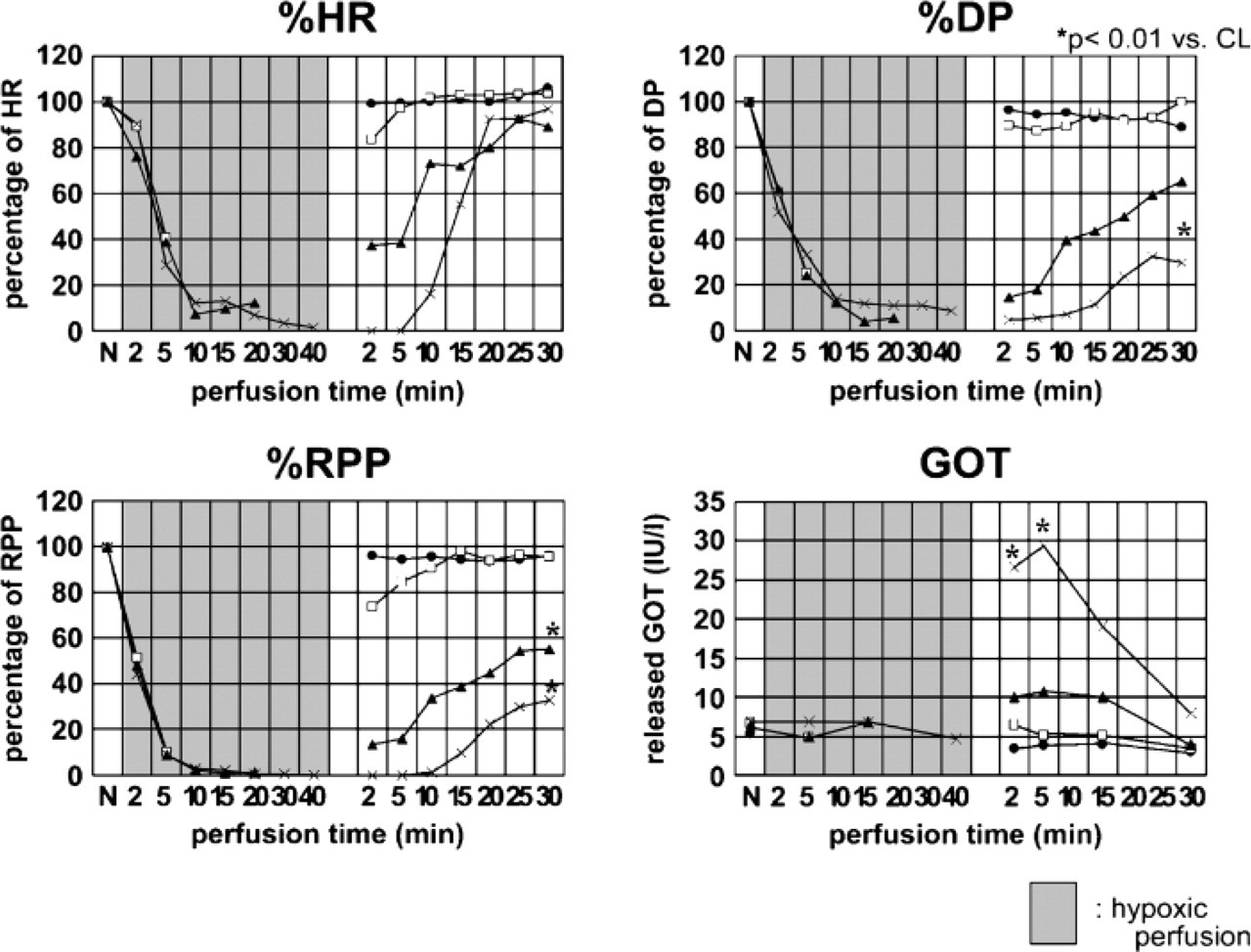
Cardiacfunctions. During experiments, %HR, %DP, and %RPP were recorded. Hemodynamic changes in hypoxia–30-min reoxygenation group are shown. These parameters were decreased during hypoxic perfusion and recovered by reoxygenation. %HR recovered to the prehypoxic level in all groups, whereas %DP and %RPP recovered to an incomplete level. The longer the hypoxic time, the lower the value of %DP and %RPP induced. Glutamic oxaloacetic trans-aminase (GOT) collected from coronary during perfusion was significantly increased in the heart that was exposed to hypoxia for 40 min at 2, 5, and 15 min of reoxygenation period (∗p<0.01) and peaked at 5 min after reoxygenation. CL, control; closed circle, CL; open square, hypoxia for 5 min; closed triangle, hypoxia for 20 min; X, hypoxia for 40 min, respectively.
The value of %DP or %RPP in the heart exposed to hypoxia for 5 min recovered to prehypoxic value. However, recovery of %DP or %RPP at the end of reoxygenation was incomplete in the heart exposed to hypoxia for 20 min or 40 min. There was a significant difference in the %DP or %RPP between CL and 20-min or 40-min hypoxia group after reoxygenation for 30 min (%DP: 5 min = 101.6 ± 9.9%, 20 min = 67.3 ± 5.5%, 40 min = 34.5 ± 6.6%∗ vs CL = 89.0 ± 3.0%, respectively, %RPP: 5 min = 95.6 ± 1.5%, 20 min = 55.2 ± 4.7%∗, 40 min = 32.9 ± 5.9%∗ vs CL = 96.1 ± 1.8%∗, respectively, ∗p<0.01 vs CL).
The released GOT was increased significantly at 2, 5, and 15 min after reoxygenation in the 40-min hypoxia group compared with CL (2 min = 26.6 ± 4.6∗ vs 4.0 ± 0.6, 5 min = 29.3 ± 5.3∗ vs 4.0 ± 0.3, 15 min = 19.1 ± 1.8∗ vs 4.0 ± 0.3: 40-min hypoxia vs CL, respectively, ∗p<0.01). The value reached the peak at 5 min after reoxygenation. The other groups did not demonstrate a significant change of GOT.
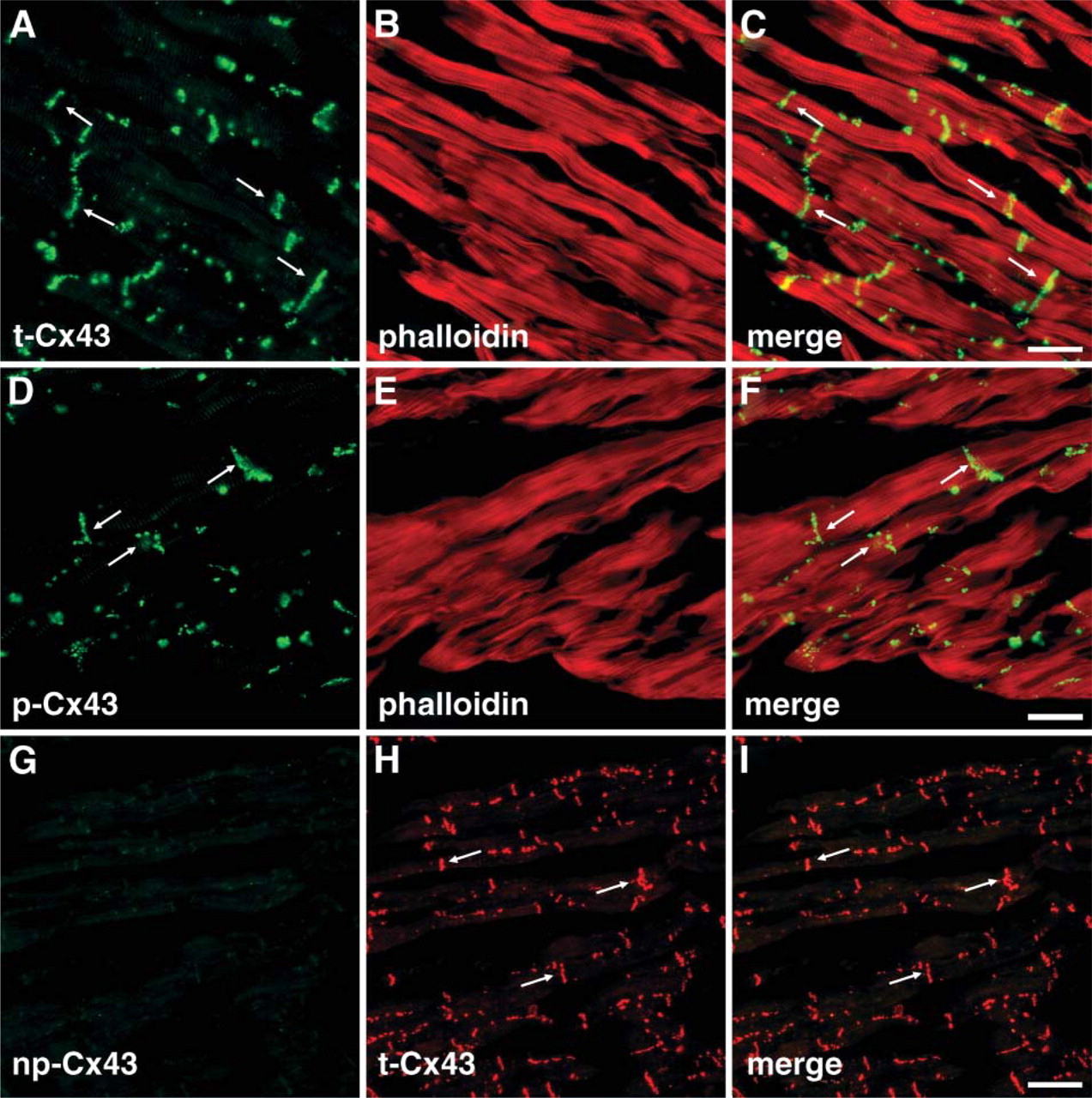
Localization of Cx43s in control heart. In control heart, double staining with anti-t-Cx43 antibody and rhodamine–phalloidin showed that Cx43 was exclusively localized at the intercalated disk (arrows) (
Immunofluorescence
In this study we used three kinds of antibodies against Cx43 for immunocytochemical and Western blot analysis (Table 1). Anti-Cx43 antibody indicating as anti-total-Cx43 (t-Cx43) antibody recognizes both phosphorylated and non-phosphorylated forms of Cx43. Anti-phospho-Cx43 antibody indicating as anti-p-Cx43 antibody especially recognizes the phosphorylated form of Cx43 at Ser368. Anti-non-phospho-Cx43 antibody indicating as anti-np-Cx43 antibody recognizes only non-phosphorylated Cx43 at Ser 368. To investigate the localization of phosphorylated and dephosphorylated Cx43, we performed double immunostaining using anti-t-Cx43 antibody and anti-p-Cx43 antibody with rhodamine–phalloidin, which is specifically bound to the filamentous actin in cardiomyocytes. In the control heart, all Cx43 recognized with both antibodies exclusively localized at the intercalated disk (Figures 33-A3). When the sections of normal heart were stained with anti-np-Cx43 antibody, no signal appeared (Figures 3G and 3H). A monoclonal antibody for detecting np-Cx43 is claimed that the antibody also recognizes some phosphorylated Cx43 in cultured cells (Cruciani and Mikalsen 1999). However, we could not observe any signals for np-Cx43 in the heart in vivo. These results indicate that Cx43 in control heart are constantly phosphorylated and located at the intercalated disk.
In the heart exposed to hypoxia, signals for np-Cx43 appeared diffusely along the non-disk region of the plasma membrane of cardiomyocytes and are somewhat heterogeneously distributed at the intercalated disk (Figure 4). Double staining with anti-t-Cx43 antibody and anti-np-Cx43 antibody showed that those signals were completely colocalized (Figure 4). Double staining with anti-p-Cx43 antibody and anti-np-Cx43 antibody showed that both phosphorylated and dephosphorylated Cx43s mingled at the intercalated disk, but that the intensity of the signal for np-Cx43 in some parts of the intercalated disk was decreased (Figures 4D–4F). In addition, the diffuse signal for Cx43 along the non-disk region was shown only for the dephosphorylated form.
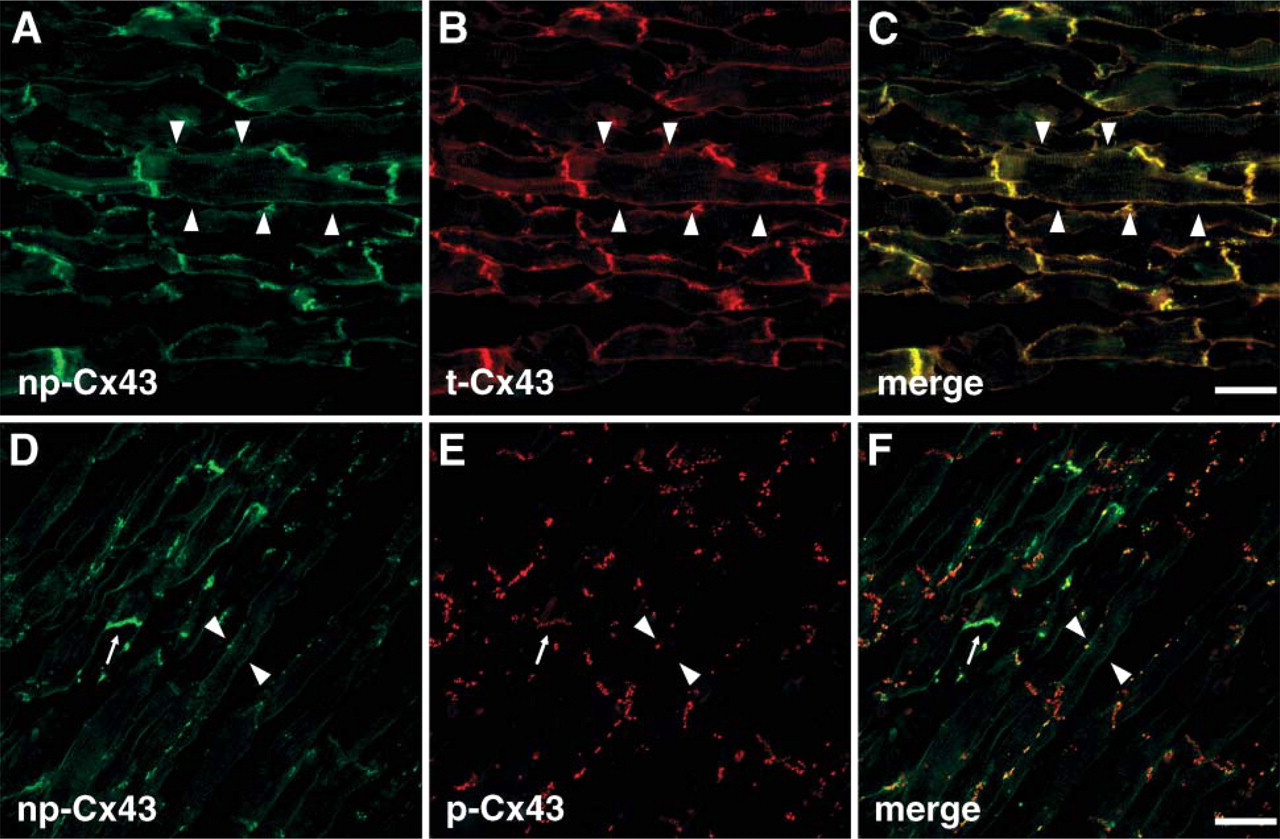
Localization of Cx43s in injured heart. In the heart exposed under hypoxic condition, double staining with anti-np-Cx43 antibody and anti-t-Cx43 antibody showed that both signals were colocalized completely (
Quantitative Analysis of the Area of Dephosphorylated Cx43
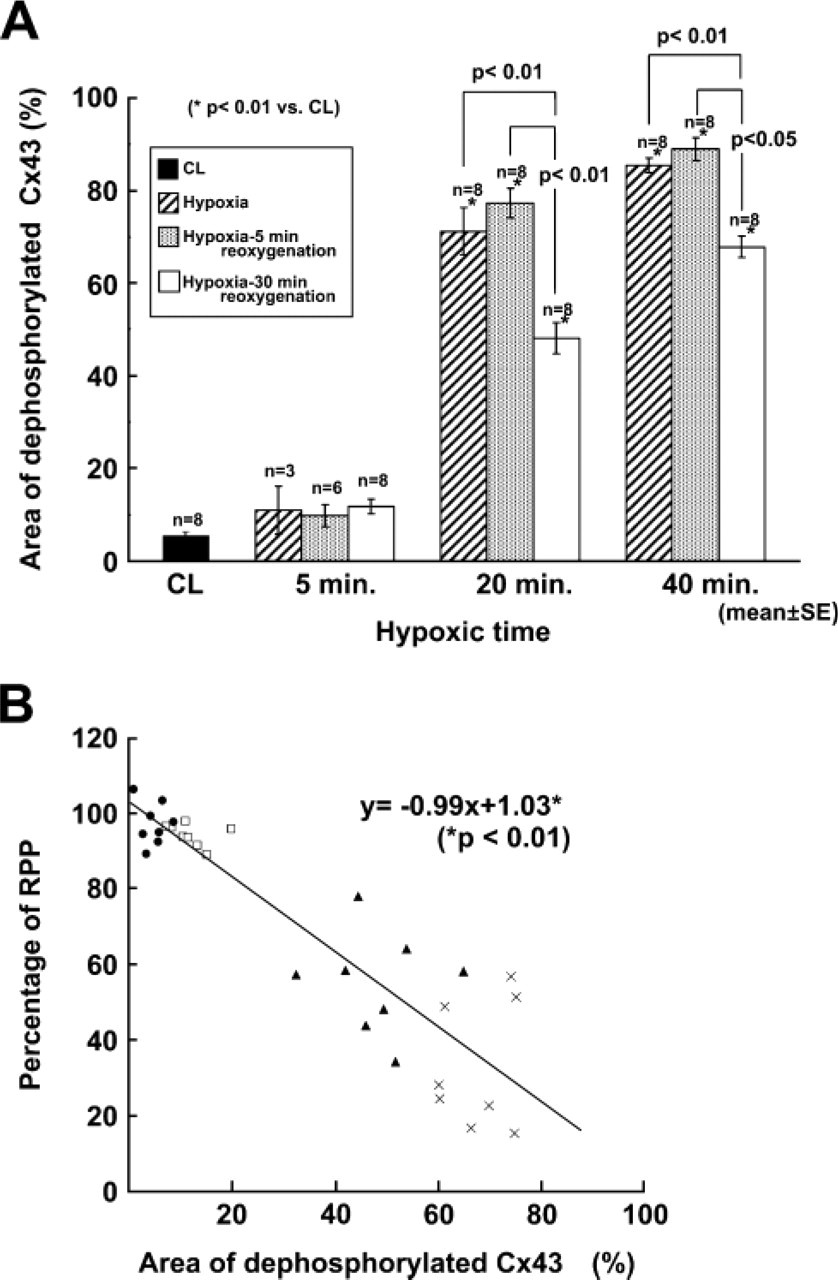
Cardiomyocytes stained with anti-np-Cx43 antibody exhibited a diffuse signal along the non-disk region of the plasma membrane. The number of cardiomyocytes stained for the non-disk region with anti-np-Cx43 antibody was increased as exposure time for hypoxia increased. To estimate the number of np-Cx43-positive cardiomyocytes, the relative area occupied by np-Cx43-positive cardiomyocytes (the dephosphorylated area) was measured on individual sections (Figure 5).
In the hypoxia–30-min reoxygenation group, the dephosphorylated area in the heart exposed to hypoxia for 20 or 40 min was significantly broader compared with that in the CL heart. However, there was no significant difference between the dephosphorylation area in the heart exposed to hypoxia for 5 min and that in CL heart (5 min = 11.7 ± 1.6%, 20 min = 48.2 ± 3.4%∗, 40 min = 67.9 ± 2.3%∗, CL = 5.3 ± 1.1%: mean ± SE, respectively, ∗p<0.01 vs CL). The area in the heart exposed to hypoxia for 40 min was significantly broader than that in the heart of 20-min hypoxia. These results revealed that the area of dephosphorylated Cx43 expanded broader as hypoxic time was longer.
In the hypoxia group there was a similarity with the hypoxia–30-min reoxygenation group at the end of the experiment. The hypoxic area also became broader in a time-dependent manner. In addition, the area of sections in the hypoxia group was significantly broader than that of the sections in the 30-min reoxygenated group following the same hypoxic time (5 min = 10.9 ± 5.1% vs 11.7 ± 1.6%, 20 min = 71.3 ± 5.1% vs 48.2 ± 3.4%∗, 40 min = 85.5 ± 1.6% vs 67.9 ± 2.3%∗; hypoxia group vs 30-min reoxygenation group, respectively, ∗p<0.01 vs hypoxia group).
As previously shown, the amount of released GOT reached the maximum level at 5 min of reoxygenation (Figure 2). The quantitative area analysis of dephosphorylated Cx43 for hypoxia–5-min reoxygenation groups showed a slight increase but no statistical significance compared with the hypoxia group (5 min = 10.9 ± 5.1% vs 9.7 ± 2.4%, 20 min = 71.3 ± 5.1% vs 79.6 ± 3.2%, 40 min = 84.7 ± 1.6% vs 88.6 ± 2.6%: hypoxia group vs 5-min–reoxygenation group, respectively).
Quantitative analysis of dephosphorylated area of Cx43. (
Image analysis indicated that dephosphorylation of Cx43 was induced mainly during hypoxia period and did not increase the dephosphorylated area during the early reoxygenation period when cardiac injury was often observed.
Correlation between Cardiac Function and Dephosphorylated Area of Cx43
Cardiac function was decreased in hypoxia in a time-dependent manner, whereas the dephosphorylated area of Cx43 was increased in a time-dependent manner. We plotted the scattergram to determine the correlation between cardiac function and dephosphorylation status of Cx43 in the 30-min reoxygenation group (Figure 5). %RPP value at the end of reoxygenation was used for cardiac function. The slope was calculated by the following formula: y = − 0.99 × + 1.03 (p<0.01). There was a statistically significant correlation between cardiac function reflected by %RPP and dephosphorylated area of Cx43.
Western Blot Analysis
To determine the quantitative change in phosphorylation status of Cx43, we performed Western blot analysis in hypoxia–reoxygenation groups (Figure 6). As previously reported (Doble et al. 2000; TenBroek et al. 2001; Barker et al. 2002), anti-t-Cx43 antibody recognized two major bands. The higher band (44–46 kDa) was the phosphorylated form and the lower band (41 kDa) was the dephosphorylated form. Anti-np-Cx43 antibody recognized only the lower band (Nagy et al. 1997). Only the higher band was detected in control heart by anti-t-Cx43 antibody. In hypoxic condition, both higher and lower bands were detected. The density of the higher band (phosphorylated form of Cx43) was decreased and the lower band (dephosphorylated form) was increased in a time-dependent manner. Immunoblot analysis with anti-p-Cx43 antibody or anti-np-Cx43 antibody showed that p-Cx43 was decreased in a time-dependent manner under hypoxic condition, whereas np-Cx43 was detected as the faint band in control heart but increased the density under hypoxic condition.
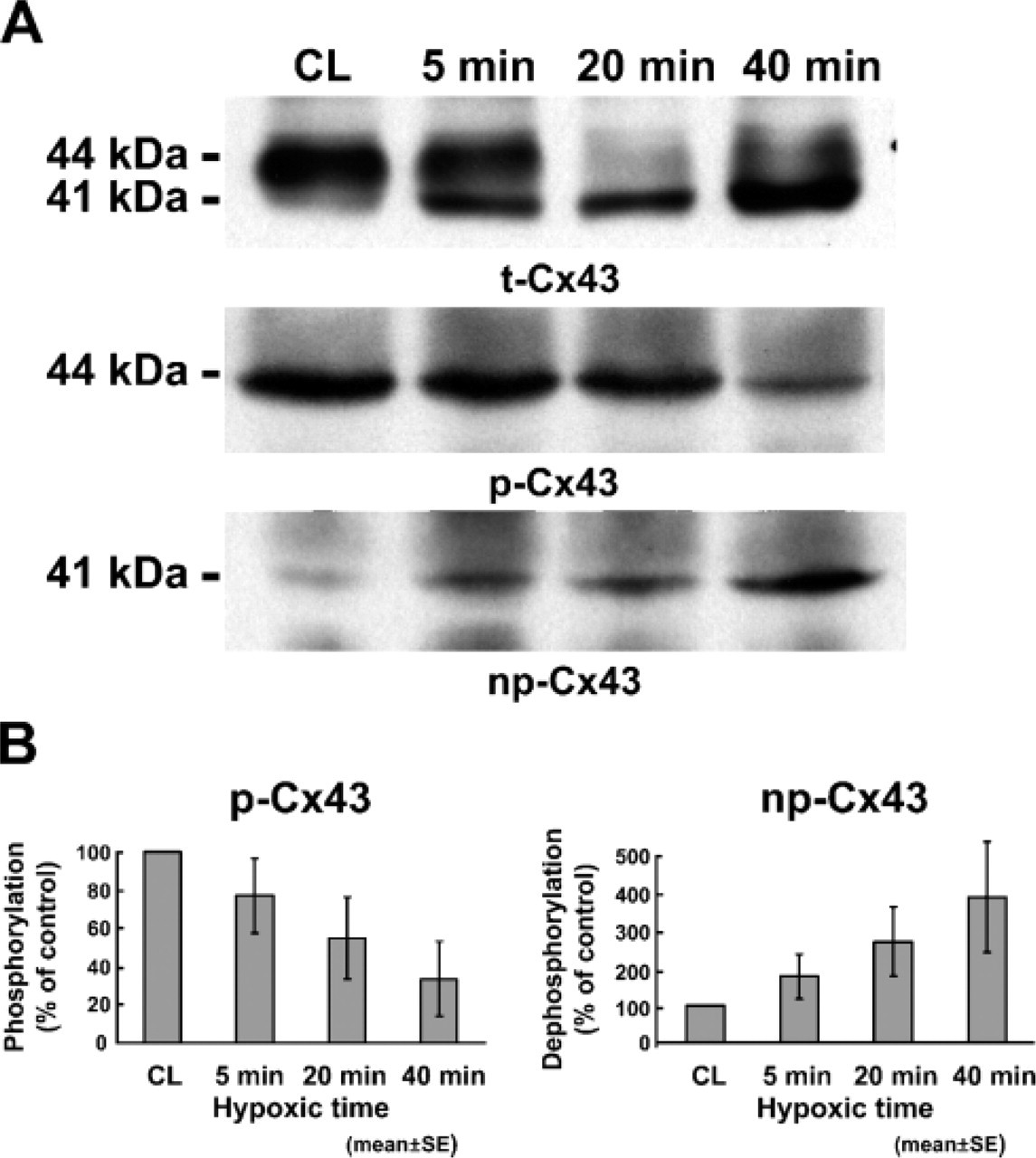
Western blot analysis. (
Localization of ZO-1 under Hypoxic Condition
It is known that ZO-1 binds directly to Cx43 at the intercalated disk of cardiomyocytes. We investigated whether or not the localization of ZO-1 was changed under the hypoxic condition. Double staining with ZO-1 and t-Cx43 showed that both proteins were colocalized at the intercalated disk in control heart (Figures 7A–7C). In the injured heart, the localization of ZO-1 was not changed, although a part of Cx43 migrated to the entire plasma membrane (Figures 7D–7F).
Discussion
In this study we investigated the relationship between phosphorylation changes of Cx43 and cardiac function under hypoxic condition in the rat heart. We clearly demonstrated that the amount of dephosphorylated Cx43, which migrated from the gap junction to the entire plasma membrane, had a positive correlation with the reduction of cardiac function reflected by %RPP in a time-dependent manner in vivo.
Distribution of Cx43 under Hypoxic Condition
In the control heart, almost all of the Cx43 in cardiomyocytes was phosphorylated and localized at the intercalated disk between neighboring cells. Previous studies also reported the phosphorylated form of Cx43 in many types of cells in vivo (Beardslee et al. 1998; Jain et al. 2003) and in vitro (Darrow et al. 1995; Beardslee et al. 2000). Cx43 is phosphorylated at multiple serine residues in vivo (Musil et al. 1990; Beardslee et al. 2000). In this study we used antibodies specifically recognizing the phosphorylation status of Cx43 at Ser368. Our findings indicated that Cx43 is constantly phosphorylated at Ser368 and assembles for gap junction formation at the intercalated disks in control cardiac myocytes in vivo.
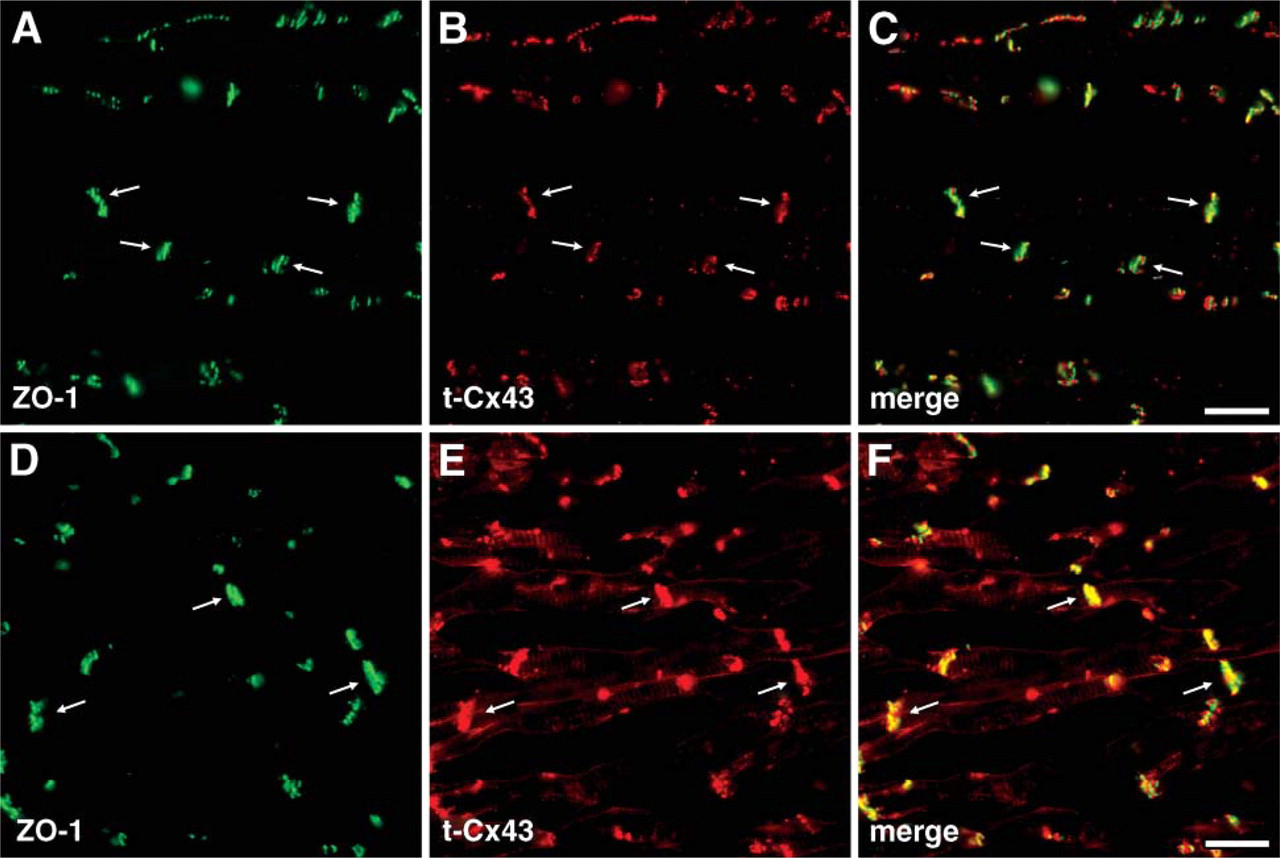
Alteration of ZO-1 under hypoxic condition. Under normal condition, ZO-1 and Cx43 were colocalized at the intercalated disk (
When the heart was exposed to hypoxia, Cx43 was dephosphorylated and redistributed to the non-disk region of the plasma membrane from the intercalated disks. Both phosphorylated and dephosphorylated Cx43s were located at the intercalated disks in hypoxic cells, but only dephosphorylated Cx43s were found on the non-disk region of the plasma membrane. Therefore, it is suggested that the dephosphorylation of Cx43 occurred at the intercalated disks and was redistributed from the intercalated disk to the non-disk region of the plasma membrane. Redistribution of Cx from the gap junction has been reported by other groups (Beardslee et al. 2000; Jeyaraman et al. 2003; Hatanaka et al. 2004; Turner et al. 2004).
Recent studies indicate that tight junction protein ZO-1 is colocalized with Cx43 at the intercalated disk (Toyofuku et al. 2001; Ferreira-Cornwell et al. 2002). The carboxyl terminal of Cx43 is directly bound to the PDZ domain of ZO-1 (Toyofuku et al. 2001; Barker et al. 2002; Duffy et al. 2004). Change of phosphorylation status of Tyr265 in Cx43 regulates the binding between Cx43 and ZO-1 (Toyofuku et al. 2001; Barker et al. 2002). Because Cx43 and ZO-1 dissociate in low pH (Duffy et al. 2004), it is possible that ZO-1 plays an important role in scaffolding Cx43 at the intercalated disk. In the present study, the dissociation between dephosphorylated Cx43 at Ser368 and ZO-1 was observed under hypoxic condition. Therefore, it is of interest to determine whether or not dephosphorylation at Ser368 of Cx43 affects the interaction between Cx43 and ZO-1.
Correlation between Cardiac Function and Phosphorylation Status of Cx43
In the present study, we showed that dephosphorylated Cx43 was increased under hypoxic condition in a time-dependent manner. Because the isolated heart was continuously perfused with low oxygenated buffer during hypoxia in our experiments, metabolites induced by low oxygen were not preserved in the tissues. However, destruction of cardiomyocytes reflected by GOT was markedly increased at the early reoxygenation injury following 40 min of hypoxia (see Figure 2), whereas dephosphorylation of Cx43 was not increased after reoxygenation. Therefore, our data suggest that the injury of hearts induced by reoxygenation is not as a consequence of the dephosphorylation of Cx43. We found a positive correlation between the area of dephosphorylated Cx43 stained with anti-np-Cx43 antibody and the %RPP. RPP value, which is calculated by HR X DP, indicates myocardial work and oxygen consumption and well reflects the whole heart function. %HR was decreased during hypoxia and increased by reoxygenation. Because %HR recovered to the prehypoxic level in all groups after 30 min of reoxygenation, %DP affects the decrease of %RPP in this study. There are many factors affecting the decrease of contraction in hypoxia or reoxygenation injury, for example, intracellular calcium concentration ([Ca2+]i) or ATP concentration. When cardiac tissue is exposed to hypoxia, [Ca2+]i is increased gradually during 9–15 min after onset of ischemia (Steenbergen et al. 1987; Jain et al. 2003), and the [Ca2+]i peaks immediately after reperfusion (Marban et al. 1990; Meissner and Morgan 1995). However, the dephosphorylated Cx43 was not increased at that point in our experiments. Therefore, it is speculated that the change of [Ca2+]i after reoxygenation does not correspond to that of dephosphorylated Cx43. According to the results, there is little possibility that [Ca2+]i regulates the dephosphorylation of Cx43. On the other hand, intracellular ATP production is rapidly decreased during ischemia and increased as oxygen is supplied (Pike et al. 1990; Meissner and Morgan et al. 1995). Beardslee et al. (2000) reported that dephosphorylation of Cx43 depends on ATP depression induced by low oxygen in vivo. Therefore, it is possible that intracellular ATP concentration may play an important role for regulating the phosphorylation status of Cx43 in vivo.
When the heart is exposed to hypoxia, the cardiomyocytes shorten to reduce contraction and increase the whole tissue resistance (Stern et al. 1985; Marban et al. 1990; Beardslee et al. 2000; Jain et al. 2003). In this study, we also recorded the end diastolic pressure (EDP) that indicates the whole tissue resistance. EDP was increased during hypoxic perfusion, peaked at the end of hypoxia, and then decreased by reoxygenation. In the heart that was exposed to hypoxia for 5 min, EDP reversed to almost the prehypoxic value at the end of reoxygenation, but in the heart exposed to hypoxia over 20 min, EDP reversed incompletely at 30 min after reoxygenation. Both the peak value and the end value were much higher in the 40-min hypoxia group than in the 20-min hypoxia group (data not shown). EDP showed similar changes as the quantitative area analysis of dephosphorylated Cx43. These data suggest that dephosphorylation of Cx43 relies on the uncoupling of cardiomyocytes that is indicated by an increase of EDP when the heart is exposed to hypoxia. In addition, our observations indicate that dephosphorylated Cx43 partially reverses to the phosphorylated Cx43 during reoxygenation. Distribution of cardiomyocytes stained with dephosphorylated Cx43 exhibited patchy, not diffuse, distribution. The patchy area containing dephosphorylated Cx43 was consistent with the severe hypoxic lesion that was indicated by pimonidazole system as a hypoxic probe. It is likely that the patchy distribution of dephosphorylated Cx43 resulted in contractile dysfunction, which is indicated as the decrease of %RPP. In addition, the patchy distribution of the “isolated” cardiomyocytes may disturb the uniform conductance front to conduct the stimuli, resulting in the cause of arrhythmia.
An important question arises whether dephosphorylation of Cx43 works for cardiac protection or for dysfunction. It is known that the intracellular concentration of hazardous metabolites such as calcium, hydrogen peroxide, and superoxide anion is increased in cardiomyocytes during hypoxia–reoxygenation injury. Gap junction closure prevents these materials from the outflow and protects the neighboring cells from damage called the “kiss of death” (Andrade-Rozental et al. 2000). It is known that preconditioning of the heart with intermittent brief ischemia protects the cardiac muscle from ischemic or reperfusion injury. In the preconditioned heart subjected to 30 min of ischemia, Cx43 was less dephosphorylated compared with the non-preconditioned controls (Jain et al. 2003; Miura et al. 2004). Their findings indicate that preservation of phosphorylated Cx43 also preserves cardiac function. Recently, it has been shown that in the preconditioned heart the ATP level before ischemia is decreased, and the rate of ATP decrease becomes slower compared with the non-preconditioned heart (Murry et al. 1990; Jennings et al. 1991; Miyamae et al. 1993). In our study, the longer the time the heart is exposed to hypoxia, the lower the recovery of the %RPP value as well as the recovery of phosphorylated Cx43 at the end of reoxygenation. These results suggest that dephosphorylation of Cx43 hampers cardiomyocytes from recovery of cardiac function in hypoxia or reoxygenation injury.
In conclusion, our present study indicates that alteration of phosphorylation and redistribution of Cx43 is an early sign of cardiac injury after hypoxia. Detection of dephosphorylated Cx43 may serve as a diagnostic tool for examining ischemic heart disease.