
Abstract
Protease-activated receptors (PARs) are multifunctional G protein–coupled receptors. Among the four existing PARs, PAR4 is preferentially expressed in the human lung tissue. However, the function of PAR4 has not been defined in the lung endothelial cells. Because PAR1-mediated cellular effects are deeply related to the morphological changes, we focused on the actin fiber and p38 mitogen-activated protein kinase (MAPK) signaling involved in actin polymerization to elucidate the role of PAR4. RT-PCR and Western blot analyses identified PAR4 expression in human pulmonary artery endothelial cells and in human microvascular endothelial cells from lung. We then examined the changes in actin fibers in endothelial cells treated with PAR4-activating peptide. PAR1-activating peptide was used for comparison. Activation of PAR4 and PAR1 by their corresponding peptides induced actin fiber formation; however, the actin filaments were broadly bundled in PAR4 as compared with the ringlike actin filaments in PAR1 activation. Correspondingly, the magnitude of p38 MAPK phosphorylation was different between cells treated with PAR4 and PAR1, with PAR4-activating peptide showing a significantly higher sensitivity to p38 MAPK inhibitor, SB203580. Taken together, these results demonstrate that activation of PAR4 results in the formation of actin fiber distinct from that by PAR1 activation, suggesting PAR4 may play specific roles in the lung endothelial cells.
Keywords
P
Stimulation of PARs is known to induce a variety of cellular effects in many types of cells. In the endothelial cells, for example, PAR1 upregulated cyclooxygenase-2 expression (Houliston et al. 2002) and intercellular adhesion molecule-1 transcription (Rahman et al. 2002), and PAR2 induced tissue factor expression and von Willebrand factor release (Langer et al. 1999). These results indicate the multifunctionality of these G protein–coupled receptors and show the functional involvement of receptors in a number of events taken place in endothelial cell. However, functional analysis of PAR4 has been limited to the studies in platelets, smooth muscle cells, and epithelial cells (Bretschneider et al. 2001; Asokananthan et al. 2002; Covic et al. 2002; Henriksen and Hanks 2002), but not in endothelial cells.
Previously, we have shown the preferential expression of PAR4 in human lung vascular endothelial cells in vitro (Fujiwara et al. 2004). In PARs signaling, activation of mitogen-activated protein kinase (MAPK) takes an important part in endothelial cell function (Marin et al. 2001; Kataoka et al. 2003). Among the MAPK family, p38 MAPK was shown to regulate actin cytoskeletal remodeling in pulmonary microvascular endothelial cells on intercellular adhesion molecule-1 ligation (Wang and Doerschuk 2001). Furthermore, remodeling of actin fibers was deeply involved in the major functions of endothelial cells, such as permeability (Kouklis et al. 2004), endothelium-dependent relaxation (Hamilton et al. 2001), cell migration (Vasanji et al. 2004), microtubele integrity (Bayless and Davis 2004), and leukocyte adherence (Vergnolle et al. 2002).
In this study, we investigated whether PAR4 and PAR1 play different roles in actin reorganization in human pulmonary artery endothelial cells (HPAEC) and human microvascular endothelial cells from lung (HMVEC-L), and whether the actin formation by elicitation of PAR4 or PAR1 is p38 MAPK-dependent in these human lung endothelial cells. Furthermore, we examined if PAR4-induced actin fibers display different morphology from the PAR1-induced actin fibers. The results indicated that the functional role of PAR4 in lung endothelial cells involved actin fiber formation and that the resulting morphology of the fibers differed from that derived from PAR1 activation through a distinct signaling pathway.
Materials and Methods
Reagents
Human α-thrombin was purchased from Sigma (St Louis, MO). The anti-PAR4 polyclonal goat antibody was from Santa Cruz (Santa Cruz, CA). The anti-p38 MAPK, anti-phospho-p38 MAPK, anti-Hsp27, and anti-phospho-Hsp27 antibodies were from Cell Signaling Technology Inc. (Beverly, MA). PAR1-activating peptide, SFLLRN-0H, was from BACHEM (Budendorf, Switzerland) and PAR4-activating peptide, GYPGQV-NH2, and its scrambled peptide, YGPGQV-NH2, were from BioGate Co., Ltd (Gifu, Japan). All the peptides used in the experiments were HPLC grade with >95% purity. Aminopeptidase inhibitor, amastatin, was from Peptide Institute, Inc. (Osaka, Japan).
Endothelial Cell Culture and Tissue Sections
HPAEC and HMVEC-L obtained from Clonetics (Walkersville, MD) were cultured in endothelial cell basal medium (EBM) (Clonetics) supplemented with endothelial growth medium-microvascular singlequots. Cells were plated onto gelatin-coated flasks, and grown under 5% CO2 at 37C.
Formalin-fixed, paraffin-embedded tissue sections from human lung and lymph node were obtained from the surgical pathology division.
Immunohistochemistry
Tissue sections (4 μm) were deparaffinized in xylene and rehydrated in ethanol series. Endogenous peroxidase activity in the sections was blocked by 0.3% (v/v) hydrogen peroxide in distilled water for 10 min. To retrieve antigens, the sections were placed in an antigen retrieval medium (Immunosaver, Nissin EM Co., Ltd, Tokyo, Japan) and heated at 98C for 1 hr. After cooling the sections to the room temperature, nonspecific binding sites were inhibited by incubation with 1:10 normal rabbit serum for 30 min. The slides were then incubated overnight with a 1:50 dilution of polyclonal goat anti-PAR4 antibody. The reaction product was visualized using the labeled streptavidin-biotin system (DAKO; Kyoto, Japan) and 3, 3'diaminobenzidine as a chromogen, and the sections were counterstained with hematoxylin. Negative control sections were processed by substitution of the primary antibody with normal goat serum. The intensity of immunostaining was semiquantitated as: -, negative; +, weak; ++, moderate; and + + +, strong.
Semiquantitative RT-PCR
Cultured endothelial cells were washed and then lysed in guanidine thiocyanate-containing buffer and total RNAs were extracted using the RNeasy Mini Kit (Qiagen; Hilden, Germany) with DNase I treatment. Semiquantitative RT-PCR for PAR4 was performed as previously described (Fujiwara et al. 2004). The PCR was performed at 94C for 45 sec, 58C for 45 sec, and 72C for 2 min. After 24 cycles of amplification, cDNA products were visualized with SYBR Green I (Molecular Probes; Eugene, OR) and band images were captured using Molecular Imager FX. Signal intensity of PAR4 was then quantitated by PDQuest software (Bio-Rad; Hercules, CA) and normalized to or against glyceraldehyde-3-phosphate dehydrogenase signal intensity. Independent experiments were conducted three times. RT-PCR reaction with no SuperScript RNase H- reverse transcriptase did not show any PCR products.
Western Blot Analysis for PAR4 and p38 MAPK Activations
Endothelial cells were plated in 60-mm dishes to reach sub-confluence. After treatments, cells were washed and lysed either in radio immuno precipitation assay buffer for PAR4 (PBS, 1% [v/v] NP-40, 0.5% [w/v] sodium deoxycholate, 0.1% [w/v] SDS, 0.1 mg/ml PMSF, 50 μg/ml aprotinin) or in buffer for p38 MAPK and Hsp27 analysis (50 mM HEPES, pH7.4, 50 mM NaCl, 5 mM EDTA, 1% [v/v] Triton X-100, 10% [v/v] glycerol, 1 mM Na3VO4, 100 mM NaF, 10 mM sodium pyrophosphate, and 34 μg/ml Aprotinin). SDS-PAGE was performed by loading samples (100 μg/lane for PAR4 and 30 μg/lane for p38 MAPK and Hsp27) in 5–15% gradient gels. Protein was transferred electrophoretically to a polyvinyl difluoride membrane for 1 hr. The membrane was incubated in TBS (10 mM Tris HCl, pH 8.0, and 150 mM NaCl) containing 10% FBS for 1 hr and with TBS containing 0.05% (v/v) Tween 20, 10% FBS, and each primary polyclonal antibody (1:100 for PAR4, 1:300 for p38 MAPK, and 1:500 for Hsp27). After three washes with TBS containing 0.05% (v/v) Tween 20, the binding of the anti-PAR4 antibody was detected with biotinylated anti-goat IgG and avidin:biotinylated enzyme complex (Vector; Burlingame, CA). Immunoreactive bands for p38 MAPK and Hsp27 were visualized by chemiluminescence (ECL Plus; Amersham Pharmacia, Piscataway, NJ) with anti-rabbit or mouse antibody-horseradish peroxidase (1:4000). Intensity of bands was analyzed by NIH Image, performed on a Macintosh computer using the public domain NIH Image program developed at the US National Institutes of Health, which were available at http://rsb.info.nih.gov/nih-image/. After quantifying the bands, signal intensities of phosphorylated p38 MAPK (pp38 MAPK) were normalized to or against p38 MAPK signal intensities and fold increase in phosphorylation was calculated.
Actin Fluorescence Staining
HPAEC and HMVEC-L (2X104 cells/1.7 cm2) were cultured in Lab-Tek II chamber slides (NUNC; Rochester, NY) coated with gelatin and grown for 3–4 days to attain confluence. Cells were serum starved for 18 hr before addition of α-thrombin (Birukova et al. 2004; Fujiwara et al. 2004), SFLLRN (Vouret-Craviari et al. 1998; Fujiwara et al. 2004), GYPGQV, or dH2O in EBM medium (0.25% [w/v] BSA) with 5 μM amastatin. After exposure to the experimental conditions for the indicated period, the cells were washed with HBSS and fixed with 10% (v/v) neutralized formalin solution and then permeabilized with 0.1% (v/v) Triton X-100. The actin filaments of cells were stained with Alexa 488-phalloidin (Molecular Probes) for 30 min at room temperature, washed three times with PBS, mounted with FluoroGuard (Bio-Rad), and examined under a confocal laser scanning microscopy equipped with X20 and X40 objective lenses (Leica; Wetzlar, Germany). Independent experiments were conducted three times.
p38 MAPK Inhibition
In the p38 MAPK inhibitory experiments, cells were first pretreated with 2.5 μM SB203580 for 15 min. At 2.5 μM of concentration, SB203580 clearly suppressed GYPGQV (PAR4 activator)-induced actin fibers and thus this dose was used in the present study. After the pretreatment, various agents were applied to the cells in the presence of SB203580 (2.5 μM) or vehicle for the indicated periods. The analyses were done by actin fluorescence staining and Western blot as described previously.
Results
Expression of PAR4 in Human Lung Tissue
To assess in vivo status of PAR4 expression in human lung, immunohistochemical staining for PAR4 was applied on lung tissues. Distinct PAR4 expression was observed in vascular endothelial cells, type II alveolar epithelial cells, and some inflammatory cells. The endothelial cells in microvessels (Figure 1A; arrow) showed a strong reaction for PAR4 similar to that in the large vessels (Figure 1B; arrow). On the other hand, PAR4 expression was not detected in lymph node tissue stained in parallel (Figure 1C), as was previously shown by Northern blot analysis (Xu et al. 1998).
Expression of PAR4 in HPAEC and HMVEC-L
By a semiquantitative RT-PCR, both HPAEC and HMVEC-L expressed PAR4 mRNA (Figure 2A), and the expression level in HMVEC-L (Figure 2A, Lane 2) showed 1.97 ± 0.01–fold increase over the expression level in HPAEC (Figure 2A, Lane 1).
Western blot analysis revealed the expression of PAR4 in both HPAEC and HMVEC-L, with more abundance in HMVEC-L (Figure 2B, Lane 2). The specificity of Western blot analysis was confirmed by detection of a weak PAR4 band in human aortic endothelial cell corresponding to its mRNA level by RT-PCR (Fujiwara et al. 2004).
PAR4 Activation and Induction of Actin Fiber Formation
When HPAEC were exposed to various concentrations of the PAR4-activating peptide, GYPGQV (20, 100, and 500 μM), for 30 min (Figure 3), only subtle effects in the width of actin bundles were observed at 20 μM and 100 μM. However, the reaction at the concentration of 500 μM resulted in the formation of dense bundles of long actin filaments. Compared with the control cells treated with vehicle, actin bundles were thick and densely localized at the cell boundary.
Exposure of GYPGQV (500 μM) for 15 min induced actin bundles at the cell boundary in HPAEC (Figure 4A) and in HMVEC-L (Figure 4B). In HPAEC, GYPGQV treatment showed increase in the thickness of actin bundles. However, the fluorescence intensity in HPAEC was not as dense as that of the 30-min treatment (see Figure 3). In HMVEC-L, exposure of GYPGQV induced dense and thickened actin fibers with strong Alexa 488–phalloidin actin staining (Figure 4B; GYPGQV, lower panels). YGPGQV (500 μM), a negative control scrambled peptide, did not alter the actin architecture in either HPAEC (Figure 4A; YGPGQV) or HMVEC-L (Figure 4B; YGPGQV), and showed similar cell cytoskeletons as in the control cells.
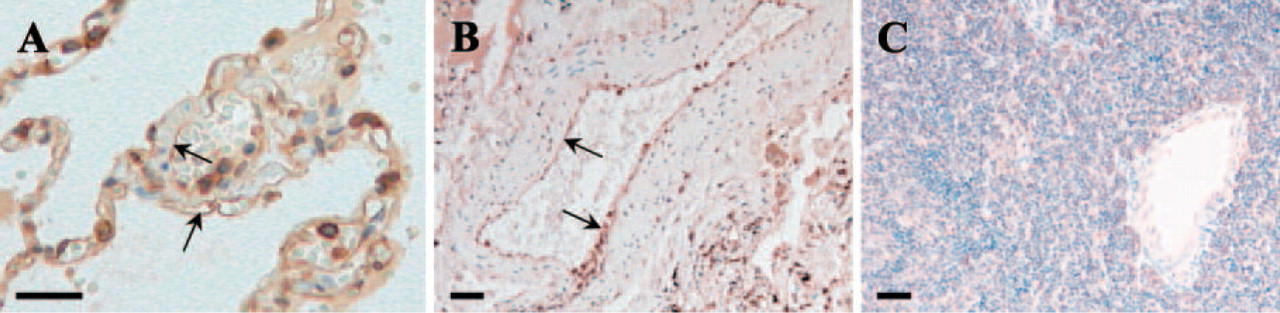
Protease-activated receptor (PAR)4 expression in human lung endothelial cells. Polyclonal anti-PAR4 antibody was used for immunohistochemical staining. (
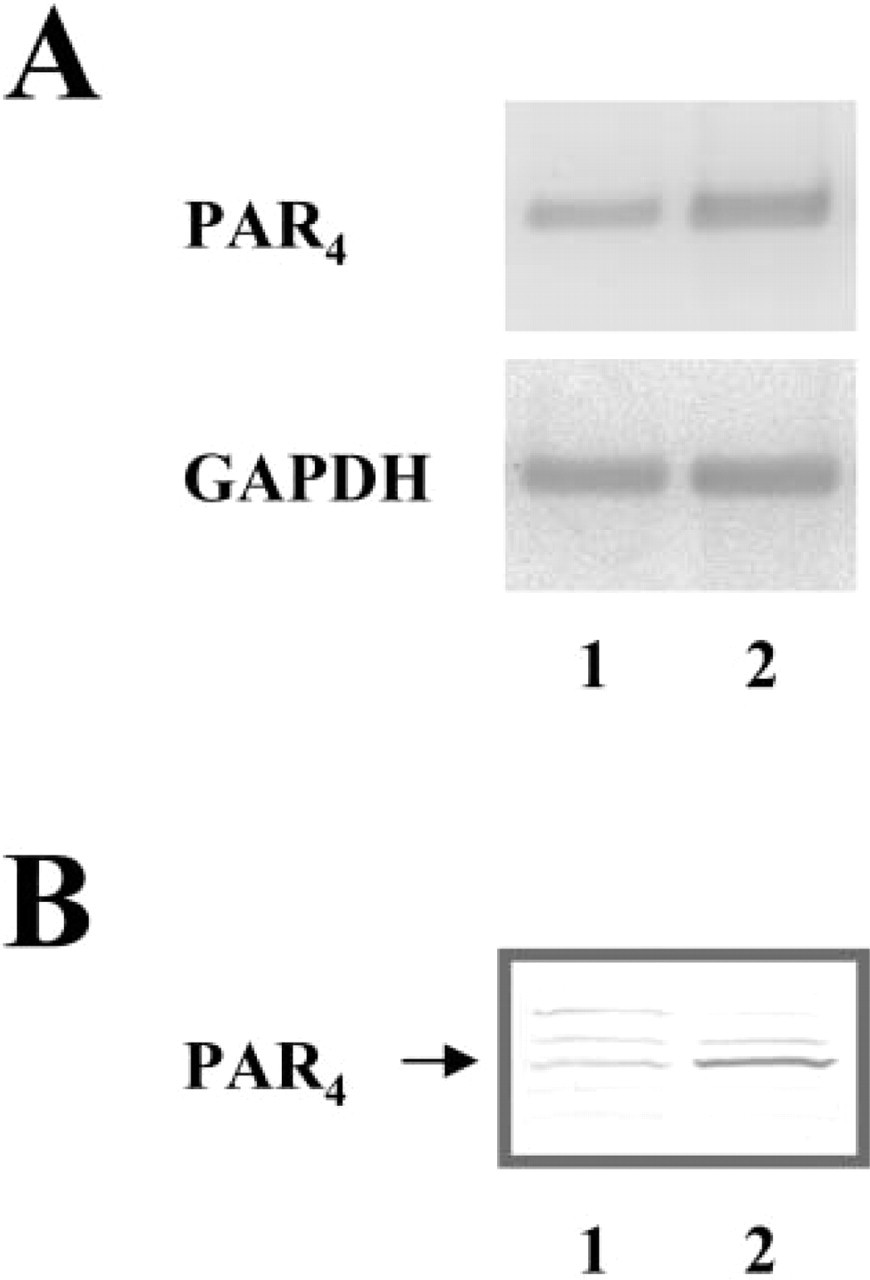
Protease-activated receptor (PAR)4 expressions in human pulmonary artery endothelial cells (HPAEC) and human microvascular endothelial cells from lung (HMVEC-L). (
Change in actin architecture was also recognized in the lung endothelial cells when the cells were treated with α-thrombin or PAR1-activating agent, SFLLRN, for 15 min (Figure 5). We used effective dose of α-thrombin (2 U/ml) or PAR1-activating peptide (100 μM) based on the results of previous studies (Vouret-Craviari et al. 1998; Fujiwara et al. 2004). Reorganized actin fiber in HPAEC (Figure 5A) and HMVEC-L (Figure 5B) was dense and formed cortical rings of polymerized actin. Furthermore, the cortical actin fiber in SFLLRN (100 μM) treated cells was similar to that in α-thrombin (2 U/ml)-treated cells. However, these cells showed different morphology from that of the cells activated with PAR4. The PAR1-activated cells were morphologically more contracted as evidenced by ringlike condensation of actin fibers that was different from actin polymerization localized at the cell boundary induced by PAR4 activation.
Involvement of p38 MAPK Phosphorylation During PAR4 Activation
Involvement of p38 MAPK, one of the MAPK members that regulate actin fiber formation, was examined in HPAEC and HMVEC-L. In HPAEC, 1.94 ± 0.11–fold increase in pp38 MAPK was detected in GYPGQV-treated cells (Figure 6A, Lane 2) than in the control cells (Figure 6A, Lane 1). Negative control peptide, YGPGQV, had no effect on p38 MAPK phosphorylation (Figure 6A, Lane 3). The SFLLRN-treated cells responded most strongly and showed 5.93 ± 0.74–fold increase in pp38 MAPK level (Figure 6A, Lane 4). Similar results were also observed in HMVEC-L (Figure 6B). Here, the phosphorylated p38 MAPK level in GYPGQV-treated cells (Figure 6B, Lane 2) was 2.45 ± 0.68–fold more than control (Figure 6B, Lane 1) or YGPGQV (Figure 6B, Lane 3)-treated cells. And SFLLRN increased pp38 MAPK level to 12.54 ± 2.81–fold (Figure 6B, Lane 4). Changes in p38 MAPK phosphorylated state were most apparent at 15 min and 5 min after the treatments for HPAEC and HMVEC-L, respectively. The expression level of total p38 MAPK was similar in both HPAEC and HMVEC-L (Figure 6, lower panel).
Suppression of PAR4-induced Actin Fiber Formation by p38 MAPK Inhibitor SB203580
To validate the role of p38 MAPK in PAR4-induced actin fiber formation, p38 MAPK inhibitor, SB203580, was used. At 2.5 μM of concentration, SB203580 suppressed GYPGQV (PAR4 activator)-induced actin fibers in HPAEC as evidenced by the decreased thickness in longitudinal axis (Figure 7A). In contrast, SB203580 could not alter actin fibers in SFLLRN (PAR1 activator) or α-thrombin–treated cells at the same concentration. In the control cells, no significant effect on actin fibers was detected under the presence of SB203580.
Activity of p38 MAPK under the presence of SB203580 was also confirmed by p38 MAPK downstream target, Hsp27 (Figure 7B). In GYPGQV-treated cells, the amount of phosphorylated Hsp27 (pHsp27) was significantly decreased under the presence of SB203580 at 2.5 μM. Similarly, 2.5 μM of SB203580 suppressed the phosphorylation state of Hsp27 in control and SFLLRN-treated cells, indicating the abolishment of MAPK activity of p38 MAPK in SB203580-treated cells. Also, the expression of Hsp27 was lower in GYPGQV-treated cells (both with and without SB203580) than control and SFLLRN-treated cells.

Actin fiber formation in human pulmonary artery endothelial cells (HPAEC) treated with various concentrations of protease-activated receptor-activating peptide, GYPGQV. HPAEC was exposed to 0, 20, 100, and 500 μM of GYPGQV in endothelial cell basal medium (0.25% [w/v] BSA). After 30 min of treatment, cells were fixed with 10% (v/v) neutralized formalin solution and stained with Alexa 488-phalloidin. Lower panels represent higher magnifications. Bars = 40 μm.
Discussion
In the present study, the expression of PAR4 and its functional roles in the lung endothelial cells were investigated. We first demonstrated PAR4 expression in the lung vascular endothelial cells with a heterogeneous expression pattern between large and small vessels. These results indicated the importance of PAR4 in endothelial cells of lung vessels, especially microvessels. Second, we demonstrated the PAR4 function that induced a distinct actin fiber formation in HPAEC and HMVEC-L. These newly formed long actin filaments with a broadened dense localization along the cell boundary were morphologically different from that of PAR1-induced cortical rings of polymerized actin. Furthermore, PAR4- and PAR1-activated cells showed a difference in terms of p38 MAPK participation, as observed by p38 MAPK inhibition experiments using SB203580. These results suggested unique capabilities of PAR4 that was not provided by PAR1, and indicated important actin-related functions of PAR4 in the lung vascular endothelial cells.
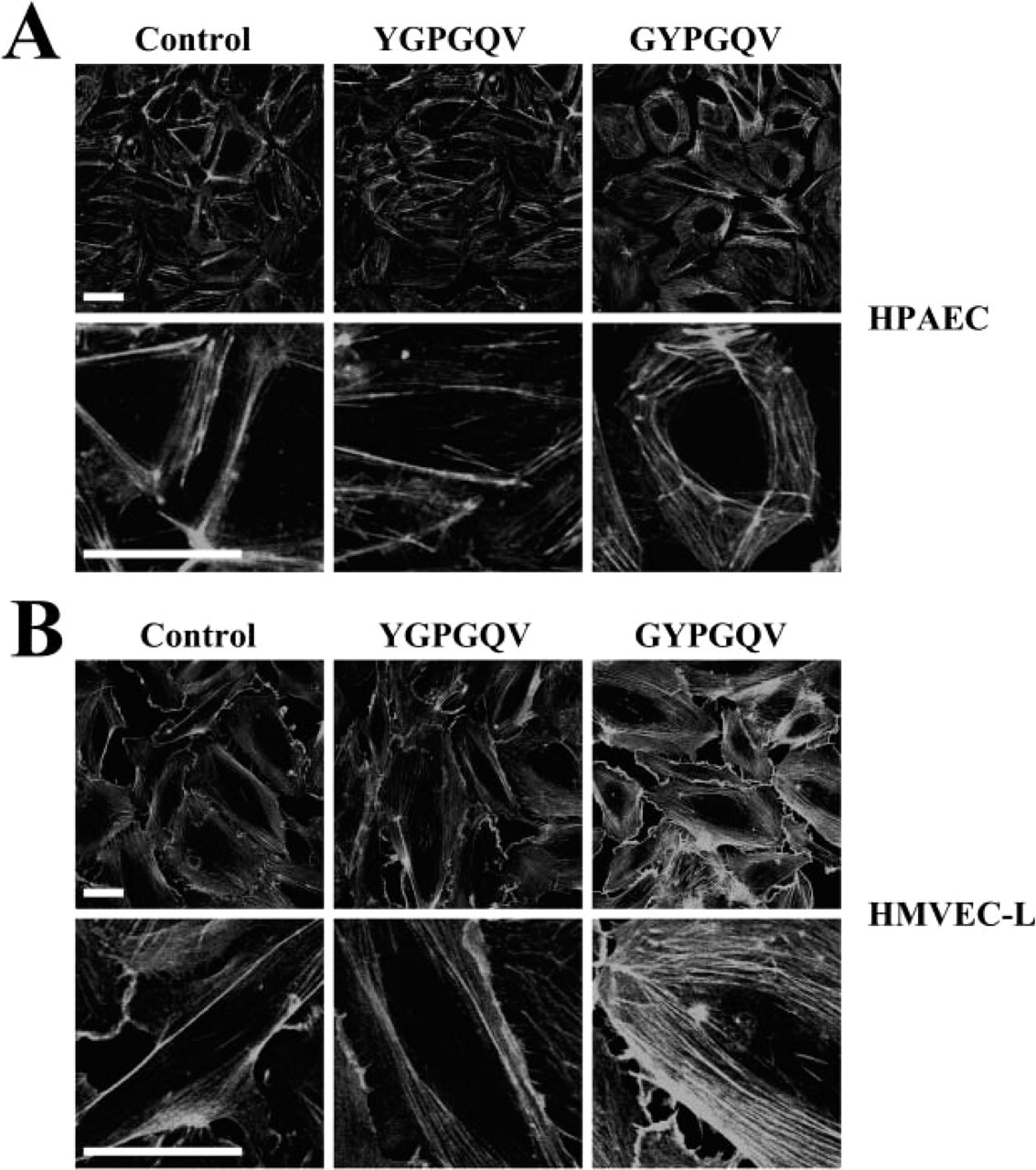
Actin fiber formation induced by GYPGQV. F-actin was detected by Alexa 488-phalloidin in human pulmonary artery endothelial cells (
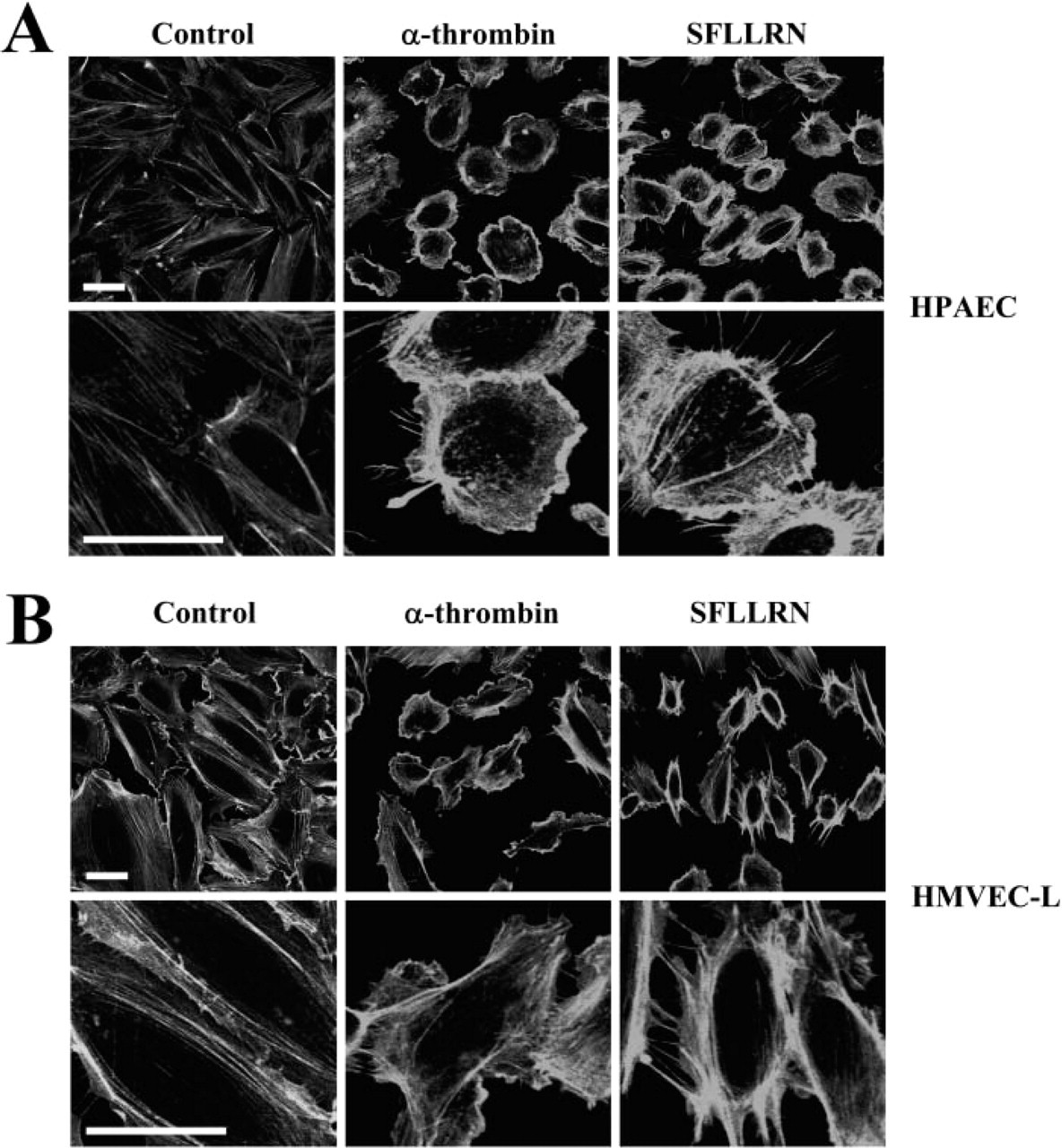
Protease-activated receptor-1 activation by α-thrombin and SFLLRN induces actin fiber formation. Human pulmonary artery endothelial cells (
As determined by immunohistochemistry, PAR4 was strongly expressed in the endothelial cells of human lung tissue, whereas lymph node tissue was unreactive. This is consistent with the previous results showing a high expression of PAR4 mRNA in human lung tissues and lack of detections in lymph node tissue (Xu et al. 1998). In addition, we found that PAR4 was also expressed in the cultured cells of HPAEC and HMVEC-L, being higher in HMVEC-L. Because our previous comparative study on PAR4 mRNA expression among human endothelial cells from pulmonary artery, aorta (Fujiwara et al. 2004), and dermal microvessel (data not shown) also showed a preferential expression of PAR4 mRNA in the lung endothelial cells, a functionally important role for PAR4 was implicated in the lung endothelial cells, especially in the capillary endothelial cells.
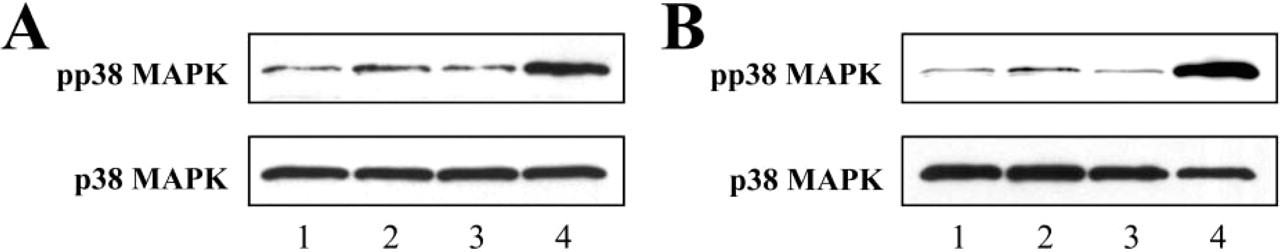
Activation of p38 MAPK in HPAEC and HMVEC-L stimulated with PAR activating peptides. (
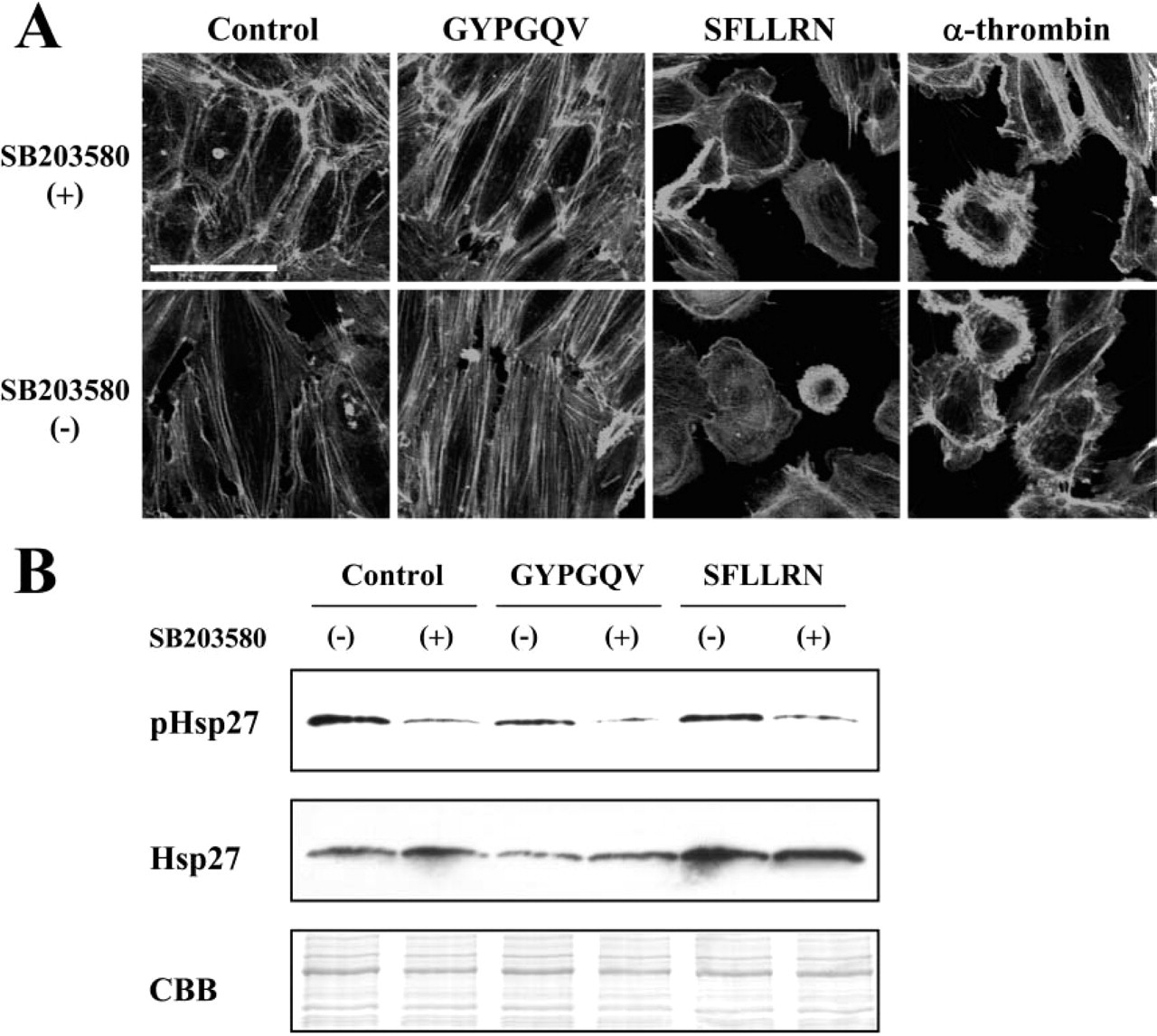
GYPGQV-induced actin fiber formation is inhibited by p38 MAPK inhibitor SB203580. (
The differences between PAR4 and PAR1 have been reported from diverse viewpoints. At the genome structure level, human PAR4 was shown to be localized at 19p12, whereas other human PARs, PAR1–3, formed a gene cluster at 5q13 (Kahn et al. 1998a). At the signaling cascade level, differential couplings of PAR1 and PAR4 to G proteins have been suggested (Faruqi et al. 2000; Asokananthan et al. 2002). Likewise, at the phenotypical level, we showed differences between PAR4- and PAR1-induced actin fiber formations. The morphology of rearranged PAR4-induced actin fibers was more broadened compared with tightened actin fibers induced by PAR1 activation. It should be noted that our study has demonstrated the morphological consequences of PAR1 activation in ECs and we have not directly measured the expression levels of PAR1 mRNA and protein in parallel, although the data could be confirmatory to our findings.
The morphological difference between PAR4 and PAR1 in rearranged actin fibers points out distinct activation kinetics of PAR4 from PAR1 in endothelial cells. Consistent with our findings, different kinetics of PAR4 were reported by others (Kahn et al. 1998b; Shapiro et al. 2000). For example, Shapiro et al. indicated that signals of PAR4 shut off more slowly than PAR1 in human platelets. Also, in the platelets, Kahn et al. indicated the requirement of higher concentration of thrombin for the activation of PAR4. This higher concentration requirement was also observed in our experiments (i.e., 500 μM of PAR4-activating peptide [GYPGQV] was needed to induce dense and thickened bundles of long filaments, whereas only 100 μM of PAR1 activating peptide [SFLLRN] was required to induce ringlike actin structures). These differences in activation kinetics indicated the unique and different roles of PAR4, which was not provided by PAR1.
The observation that PAR4 induced actin fiber formation was highly sensitive to p38 MAPK inhibitor, SB203580, raised a hypothesis that p38 MAPK could be the principal factor that controls the diverse PAR actin traffic pathways. In cardiomyocytes, PAR4 was shown to activate p38 MAPK via Src, an important upstream signaling factor for actin polymerization, whereas PAR1 was unable to activate Src, indicating the involvement of a different signal activation cascade in p38 MAPK (Sabri et al. 2003). Additionally, for example, PAR1-activated p38 MAPK was reported to induce cell proliferation in microglia (Suo et al. 2002) and in smooth muscle cells (Ghosh et al. 2002), showing MAPK activity toward the cell proliferation. These differences in PAR-p38 MAPK signaling events may explain the differences in PAR4- and PAR1-induced actin fiber formation. Interestingly, expression level of Hsp27, a factor downstream of p38 MAPK, was suppressed in GYPGQV-treated cells, whereas control and SFLLRN-treated cells showed similar expression of Hsp27. Thus our results strongly implied a distinct role for PAR4 in lung microvascular endothelial cells represented by alveolar capillaries.
Similar forms of PAR4-induced actin fibers were reported under such stimuli as mechanical stretch (Birukov et al. 2003), intercellular adhesion molecule-1 cross-linking (Wang and Doerschuk 2001), and vascular endothelial growth factor treatments (Rousseau et al. 1997), which exhibited actin fibers with thick bundles in endothelial cells. The actin fibers formed by these stimuli were involved in cell barrier, neutrophil adherence, and cell migration, respectively. Concurrently, the cytoskeletal remodeling was revealed to be p38 MAPK-dependent in these experiments, reinforcing a link between PAR4/p38 MAPK-induced actin fibers and the physiological events. These evidences further indicate the importance of PAR4 activation in endothelial cell functions, although quantitative morphometric analysis of actin remodeling in EC cultures, preferably by image analysis, could lend support to our findings.
The importance of PAR4 in lung endothelial cells could be hypothesized from the fact that PAR4 expression is upregulated in response to inflammatory stimuli, tumor necrosis factor-α, and interleukin-1α (Hamilton et al. 2001). These factors are highly expressed and are central mediators in the pathogenesis during pulmonary fibrosis (Raines et al. 1989; Piguet et al. 1993). Thus PAR4 might participate, at least in part, in the induction of pulmonary diseases. In these processes, PAR4-induced actin fiber may play significant roles in permeability control (Kiemer et al. 2002), neutrophil migration (Rousseau et al. 1997), adherence (Wang and Doerschuk 2001), and vascular relaxation (Hamilton et al. 2001).
In conclusion, we have demonstrated the expression of PAR4 in lung vascular endothelial cells and its functional effect on actin fiber formation. The morphology of PAR4-induced actin fiber was distinct from that of PAR1-induced actin fiber. The PAR4-induced actin fiber formation was highly p38 MAPK-dependent, whereas the inhibition of p38 MAPK had little effect on PAR1-induced actin formation. These results indicated that PAR4 might provide unique capabilities that could not be contributed by PAR1. Further attempts will be required to elucidate the physiological and pathological role of PAR4 in the lung vascular endothelial cells.
Footnotes
Acknowledgements
Supported by grants from the Ministry of Education, Science, Culture, and Sport of Japan and, in part, by a Maruyama Memorial Research Grant, Nippon Medical School, Tokyo, Japan. We wish to acknowledge Seiko Egawa, Department of Molecular Pathology, Institute of Gerontology, Nippon Medical School, for her assistance with laboratory analyses.