
Abstract
β2-Adrenergic agonists stimulate alveolar epithelial sodium (Na+) transport and lung fluid clearance. Alveolar type II (AT2) cells have been reported to express β2-adrenergic receptors (β2AR). Given the large surface area covered by alveolar type I (AT1) cells and their potential role in alveolar fluid removal, we were interested in learning if AT1 cells express β2AR as well. Because β2AR is potentially susceptible to desensitization by G-protein-coupled receptor kinase 2 (GRK2), we also undertook localization of GRK2. β2AR and GRK2 expression was evaluated in whole lung, isolated alveolar epithelial cells (AECs), and AECs in primary culture, and was localized to specific AEC phenotypes by immunofluorescence techniques. β2AR is highly expressed in AT1 cells. β2AR mRNA increases with time in culture as AT2 cells transdifferentiate towards the AT1 cell phenotype. Immunoreactive GRK2 is seen in both AT1 and AT2 cells in similar amounts. These data suggest that both AT1 and AT2 cells may contribute to the increased alveolar Na+ and water clearance observed after exposure to β2 adrenergic agents. Both cell types also express GRK2, suggesting that both may undergo desensitization of β2AR with subsequent decline in the stimulatory effects of β2-adrenergic agonists over time.
C
Catecholamines and agents that are selective for the β2-adrenergic receptor (β2AR) stimulate transepithelial Na+ transport in the lung under normal and pathological conditions (Charron et al. 1999; Frank et al. 2000; Saldias et al. 2000). Increases in alveolar fluid clearance are associated with high endogenous plasma catecholamine levels in such diverse conditions as septic shock in rats (Pittet et al. 1994) and neurogenic pulmonary edema in dogs (Lane et al. 1998). Exogenously-administered β adrenergic agonists have similarly led to accelerated clearance of edema fluid in rats with increased left atrial pressure (Azzam et al. 2001; Frank et al. 2000), after hyperoxic exposure (Saldias et al. 1999), and after ventilator-associated lung injury (Saldias et al. 2000). Adenovirus-mediated overexpression of β2AR in normal rats increased clearance of alveolar fluid when measured in an isolated lung model (Dumasius et al. 2001). Recently, investigators (Sartori et al. 2002a) demonstrated that inhalation of a β2-adrenergic agonist, salmeterol, attenuated development of high-altitude pulmonary edema in human subjects predisposed to that condition. Therefore, there is considerable experimental evidence to suggest that β2-adrenergic agonists may be useful in accelerating clearance of pulmonary edema in human patients.
Despite these promising results, a potential concern is that continued stimulation of β2ARs by endogenous catecholamines or by drugs that stimulate β2ARs might lead to decreased responsiveness to the beneficial effects of these agents over time (Liggett 1997). β2AR signaling occurs via agonist binding to the receptor, which then interacts with a membrane-bound G-protein. Desensitization, defined as a decline in cell response despite the continued presence of a stimulus of constant intensity, is a feature of G-protein-coupled receptors (GPCRs), such as β2AR. Decreased responsiveness of GPCRs can occur by several mechanisms, including receptor phosphorylation (mediated by cAMP-dependent protein kinase A or by non-cAMP-dependent G-protein-coupled receptor kinases; GRKs), sequestration or internalization of receptor, or decreasing receptor number (Liggett 1999; Ruiz-Gomez and Mayor 1997). GRKs are of particular interest because their expression is altered in certain disease states, such as congestive heart failure (Lefkowitz et al. 2000), hypothyroidism (Penela et al. 2001), and cystic fibrosis (Mak et al. 2002). An important GRK for desensitization of β2ARs appears to be G-protein receptor kinase 2 (GRK2), also called β-adrenergic receptor kinase 1 (βARK1) (Liggett 1997; Ruiz-Gomez and Mayor 1997). It is possible that GRK2 plays an active role in regulation of responsiveness of alveolar epithelium to β2-adrenergic agents over time.
AT2 cells have been reported to express β2AR (Fabisiak et al. 1987). Given the large surface area covered by AT1 cells and their potential contribution to alveolar Na+ and water removal, we were interested in learning if AT1 cells also express β2ARs. Here we used immunofluorescence (IF) techniques in isolated AT1 and AT2 cells, in alveolar epithelial cells (AECs) in primary culture, and in whole lung to localize β2AR and GRK2 within the alveolar epithelium of normal rat lung. We further evaluated expression of β2ARs and GRK2 in freshly isolated AT1 and AT2 cells, compared with AT2 cells in primary culture during transdifferentiation towards the AT1 cell phenotype.
Materials and Methods
Cell Isolation and Culture
This study was approved by the Institutional Animal Care and Use Committee of the University of Southern California. AECs were isolated using modifications of previously described methods for AT2 and AT1 cell isolation (Dobbs 1990; Borok et al. 1994). Briefly, AT2 cells were isolated from adult male Sprague-Dawley rats (125–150 g) by enzymatic dispersion with elastase (Worthington, Freehold, NJ; 2.5 U/ml) followed by panning on IgG-coated bacteriological plates. The enriched AT2 cell preparation (>90% purity as assessed by tannic acid staining) was resuspended in defined serum-free medium (MDSF) consisting of Dulbecco's modified Eagle's medium and Ham's F-12 nutrient mixture in a 1:1 ratio (Sigma Chemical; St Louis, MO) supplemented with bovine serum albumin (BSA; Collaborative Research, Bedford, MA), HEPES, non-essential amino acids, glutamine, penicillin G, and streptomycin (Borok et al. 1994). Cell viability was >90% as determined by trypan blue exclusion. Cells were seeded onto tissue culture-treated polycarbonate (Nuclepore) filter cups (Transwell; Corning Costar, Cambridge, MA) at a density of 1 × 106/cm2 and maintained in a humidified 5% CO2 incubator at 37C. Media were changed on the second day after plating and every other day thereafter. Cultured cells were analyzed by Northern and Western blotting at days 0, 1, 6, and 8 after plating. Cells at days 0 and 1 represent AT2 cells, while cells on days 5–8 have undergone transdifferentiation towards the AT1 cell phenotype, as assessed by morphology and expression of AT2 and AT1 cell-specific probes (Cheek et al. 1989; Danto et al. 1992; Borok et al. 1994).
To obtain freshly isolated preparations partially enriched for AT1 cells, lungs from adult male Sprague-Dawley rats (300 g) were digested with 8 U/ml elastase and 10 mg/ml collagenase type 1 (Worthington), chopped, and filtered through 100 μM strainers. Approximately 20% of the cells obtained in this fashion are AT1 cells, as determined by immunoreactivity with the mouse monoclonal Ab (MAb) VIIIB2, a marker for rat AT1 cells (Danto et al. 1992) (see below). Cytocentrifuged preparations were processed for IF after fixation in 100% cold methanol for 10 min.
Processing of Lung Tissue and Cultured Cells for IF
Normal rat lung tissue was inflated and fixed in 4% paraformaldehyde. After embedding in paraffin, samples were cut into 4-μm sections and placed on glass slides. AT2 cells were grown on polycarbonate filters. At day 5, cells on filters were fixed in 4% paraformaldehyde, washed in Tris-buffered saline (TBS, pH 7.5), and prepared for IF studies as described below.
Antibodies
The anti-β2-adrenergic receptor (β2AR) Ab is an affinity purified rabbit polyclonal Ab developed against a peptide from the carboxy terminus of mouse β2AR (Santa Cruz Biotechnology; Santa Cruz, CA). The manufacturer reports that this Ab reacts with β2ARs and β3ARs of mouse and rat origin but does not react with β1ARs. β3AR has not been previously localized to lung tissue (Evans et al. 1996; Mak et al. 1996; Sartori et al. 2002b). VIIIB2 is a mouse MAb that recognizes an epitope in the apical membrane of AT1 cells (Danto et al. 1992). Anti-GRK2 (= βARK1) Ab is a rabbit polyclonal Ab raised against a peptide from the carboxy terminus of human GRK2 that crossreacts with rat GRK2 (Santa Cruz). Normal rabbit IgG (Vector Laboratories; Burlingame, CA) was used a control for primary rabbit Abs at the same concentrations as the primary Abs. For Western blotting analysis, a mouse MAb to GRK2 was used (Santa Cruz). This MAb was raised against a recombinant peptide of human GRK2 origin and specifically reacts with rat GRK2. Polyclonal goat anti-surfactant protein C (SP-C) Ab (Research Diagnostics; Flanders, NJ) was used as an AT2 cell marker.
Immunofluorescence
Whole Lung. After deparaffinization and rehydration through graded alcohols, lung tissue sections on slides underwent microwave antigen retrieval (Antigen Unmasking Solution; Vector). Slides were then sequentially double labeled with goat anti-SP-C (1:75) and either rabbit anti-β2AR (1:300) or anti-GRK2 (1:300) Abs, or with normal rabbit IgG as a negative control. Anti-SP-C Ab was amplified using an avidin-biotin system with fluorescein isothiocyanate (FITC; Vector). After an avidin-biotin blocking step, anti-β2AR Ab or anti-GRK2 Ab was amplified using an avidin–biotin system with Texas Red (Vector). Tissue sections were treated with Vectashield Mounting Medium (Vector) containing 4′,6 diamidino-2-phenylindole (DAPI), which counterstains nuclei blue.
AECs in Primary Culture. Paraformaldehyde-fixed AECs grown on polycarbonate filters were washed in TBS with 0.05% Tween-20 (TBS-T), treated with CAS block (Zymed Laboratories, South San Francisco, CA), incubated with rabbit anti-β2AR Ab, anti-GRK2 Ab, or normal rabbit IgG overnight at 4C, and washed in TBS-T. Signal was amplified using biotinylated anti-rabbit secondary Ab (Vector), washed in TBS-T, and labeled with avidin-linked FITC (Vector). Cells on filters were treated with Vectashield containing propidium iodide, which counterstains nuclei red, and mounted on glass slides.
Freshly Isolated AECs. Methanol-fixed cytocentrifuged cells on slides were blocked in PBS, pH 7.2, with 3% BSA overnight, incubated with primary Abs for 1 hr at room temperature, rinsed in PBS, incubated with secondary Ab linked to either rhodamine (red) or FITC (green), and rinsed in PBS. Cells were identified by double labeling with AT1 or AT2 cell markers and Abs to β2AR or GRK2. To minimize cross-reactivity of primary Abs, Ab combinations were chosen so that both Abs were taken from different animal species. Slides were treated with Vectashield containing DAPI.
Slides were viewed using an Olympus BX60 microscope equipped with epifluorescence optics. Images were captured separately using a cooled CCD camera (Magnafire; Olympus, Lake Success, NY) with filters for DAPI, FITC, or rhodamine/Texas Red. The images were merged and imported into Adobe Photoshop (Adobe Systems; Mountain View, CA) as TIFF files. Where indicated, slides were also viewed with a confocal scanning laser microscope (Nikon Eclipse, TE 300; Melville, NY) equipped with an argon blue laser (excites at 488 nm) and an HeNe green laser (excites at 543 nm). Confocal images were processed with a Nikon PCM2000 laser scanner.
RNA Isolation and Northern Analysis
Total RNA was isolated from cultured AECs by the acid phenol-guanidinium-chloroform method (Chomczynski and Sacchi 1987). Equal amounts of RNA (5 or 10 μg) were denatured with formaldehyde, size-fractionated by agarose gel electrophoresis under denaturing conditions, and transferred to nylon membranes. RNA was immobilized by UV cross-linking. Hybridization was performed in hybridization buffer for 16 hr at 65C. Blots were probed with a 1.8-kb cDNA probe for rat β2AR (from American Type Culture Collection; Manassas, VA) labeled with [α32P]-dCTP (Roche Molecular Biochemicals; Indianapolis, IN) or biotin (Kirkegaard & Perry Laboratories; Gaithersburg, MD) using the random primer method. Blots were washed at high stringency (0.5 × SSC-0.1% SDS at 55C). Differences in RNA loading were normalized using a 24-mer oligonucleotide probe for 18S rRNA end-labeled with [32P]-ATP. Signal intensity was quantified using a Storm Phosphorimager (Bio-Rad; Hercules, CA).
Western Analysis
Freshly isolated AT2 cells and AECs in culture on days 6–8 were solubilized in 2% SDS sample buffer. GRK2 expression was similar in AT1-like cells at days 6 or greater in culture, so results were pooled for analysis. Protein concentrations were determined with the Bio-Rad DC Protein Assay kit. Commercially available control sample for GRK2 (Ramos cell lysate; Santa Cruz) was loaded with the AEC samples. Equal amounts of protein in sample buffer were resolved by SDS-PAGE and transferred to Immobilon-P membranes (Millipore; Marlborough, MA). Membranes were blocked in 5% non-fat dry milk in TBS-T overnight before incubation with the mouse anti-GRK2 Ab. After washing, membranes were incubated with anti-mouse IgG Ab conjugated to horseradish peroxidase (Promega; Madison, WI). Complexes were visualized using enhanced chemiluminescence (ECL; Amersham, Arlington Heights, IL). Signal intensity was quantified using the Fluorchem Imaging System (Alpha Innotech; San Leandro, CA).
Statistical Analysis
Results are expressed as mean ± SEM. Significance of differences (p<0.05) was determined by Students's t-test or, where multiple time points were compared, by one-way ANOVA with the Tukey–Kramer adjustment for multiple comparisons.
Results
Immunolocalization of β2AR
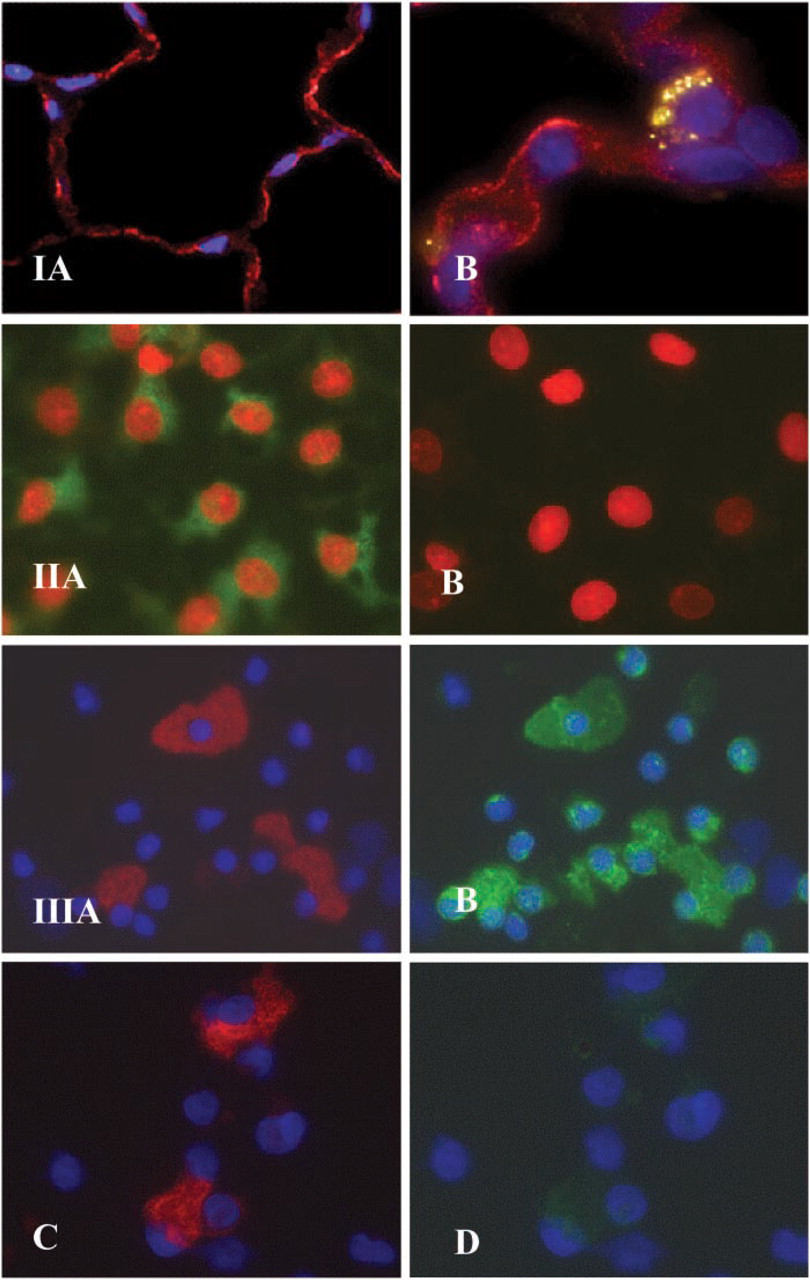
Immunoreactive β2AR was found in AECs diffusely lining the airspace in paraformaldehyde-fixed whole lung tissue (Figure 1-IA, red). β2AR was localized in the alveolar wall to thin, elongated cells with the morphologic appearance of AT1 cells, and to cuboidal cells that appear to be AT2 cells. The pattern of staining was both membrane-associated (linear) and cytoplasmic (granular). In lung tissue double labeled with anti-β2AR and anti-SP-C Abs, it was apparent at higher power (×100) that the β2AR staining was not only co-localized with staining for SP-C (Figure 1-IB, yellow-green), a marker of AT2 cells, but was also detected in adjacent cells, supporting the localization of β2AR to both AT1 and AT2 cells. Cell nuclei were identified using DAPI (blue).
AECs grown in primary culture for 5 days trans-differentiate towards the AT1 cell phenotype. These AT1-like cells were immunoreactive to anti-β2AR Ab, as shown in Figure 1-IIA. By comparison, these cultured cells were non-reactive with control rabbit IgG (Figure 1–IIB). Cell nuclei were identified using propidium iodide (red).
(
To further characterize β2AR expression in AECs, peripheral rat lung cells were enzymatically dispersed and isolated cells cytocentrifuged onto glass slides. Cytocentrifuged cell preparations were concurrently labeled with anti-β2AR Ab and a primary Ab specific for AT1 cells, VIIIB2. AT1 cells, identified by reactivity to VIIIB2 (Figure 1-IIIA, red), were immunoreactive to anti-β2AR Ab (Figure 1-IIIB, green). Other cells in the crude lung cell mixture also reacted with the β2AR antibody, including small cells that appear morphologically to be AT2 cells. No reactivity was seen when control rabbit IgG was substituted for rabbit anti-β2AR Ab in double-labeling experiments (Figure 1–IIIC and IIID). Cell nuclei were identified using DAPI (blue).
β2AR Expression in AECs in Primary Culture
As shown in this representative Northern blot, freshly isolated AT2 cells and AEC grown on polycarbonate filters for one and eight days express β2AR mRNA (Figure 2A). Densitometric analysis of three separate preparations (Figure 2B) demonstrated a 40% increase in β2AR mRNA from day 0 to day 8 (p<0.05) when corrected for levels of 18S rRNA. These findings indicate increased expression of β2AR mRNA as cells transdifferentiate from the AT2 to the AT1 cell pheno-type.
Immunolocalization and Expression of GRK2 Protein
Confocal microscopy was used to optimize localization of GRK2 in alveolar epithelium in whole lung tissue. Double labeling with anti-GRK2 and anti-SP-C Abs demonstrated that AECs express immunoreactive GRK2 (red) both with and without co-localization with SP-C (green), an AT2 cell marker (Figure 3A). This is consistent with expression of GRK2 in both AT1 and AT2 cells. GRK2 expression was most intense on the cell membrane, although some cytoplasmic staining was also seen. Substitution of normal rabbit IgG for anti-GRK2 Ab yielded no specific staining (Figure 3B). AECs were grown in primary culture for 5 days as described above for β2AR studies. These AT1-like cells were strongly immunoreactive with anti-GRK2 Ab (green), as shown in Figure 3C. By comparison, these cells were non-reactive with control rabbit IgG (Figure 3D). Cell nuclei were identified using propidium iodide (red). Isolated alveolar cells were double labeled with anti-GRK2 Ab (Figure 3E) and VIIIB2 (Figure 3F). AT1 cells that were labeled with VIIIB2 (red) also expressed GRK2 (green). Other cells present in this enzymatically dispersed lung cell preparation are probably AT2 cells or leukocytes, as has been described previously (Borok et al. 2002).
Western analysis was performed on freshly isolated AT2 cells and AT1-like cells on days 6–8 in culture to evaluate expression of GRK2 protein as a function of time. Ramos cell lysate, which has abundant GRK2 protein, was used as a positive control. As shown in this representative Western blot (Figure 4A), similar amounts of GRK2 protein were found in freshly isolated AT2 cells and AT1-like cells on day 8 in culture. Densitometry from Western blotting analyses (n = 3) is summarized in Figure 4B. The bar graph shows average densitometric values (±SEM) relative to GRK2 protein abundance on d.0 (= 100%). There was no significant difference in GRK2 expression between day 0 and days 6–8 (p = 0.58).
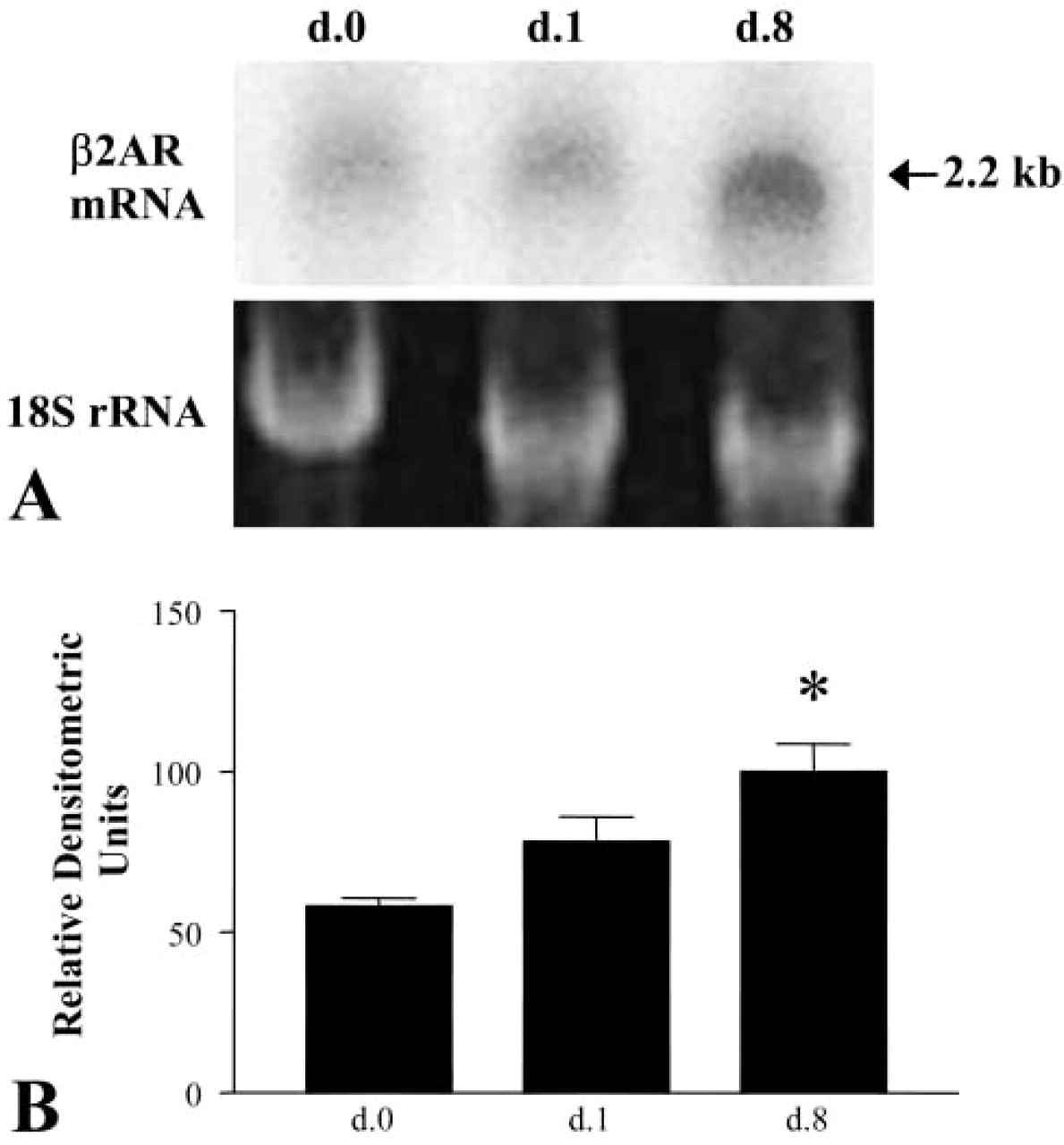
Expression of β2AR mRNA by AECs. Northern blotting analysis was performed using total RNA from purified freshly isolated AT2 cells (d.0) and AECs on day 1 (d.1) and day 8 (d.8) in culture. (
Discussion
In this study we used freshly isolated distal lung cells, AECs in primary culture, and paraformaldehyde-fixed whole lung tissue to demonstrate the presence of immunoreactive β2AR in AT1 and AT2 cells in normal rats. We also found that mRNA for β2AR was more strongly expressed in AECs that had transdifferentiated towards the AT1 cell-like phenotype in culture than in freshly isolated AT2 cells. In addition, we immunolocalized GRK2 (also called βARK1), one of the more abundant and broadly expressed G-protein receptor kinases, to both AT1 and AT2 cells in whole lung tissue and to isolated AT1 cells and AT1-like cells in culture. These data indicate that both AT1 and AT2 cells express β2AR and GRK2. Therefore, both types of cell have the capacity to respond to β2-adrenergic agonists and to modify their responsiveness in the presence of continued agonist stimulation.
Our laboratory and others have shown that active Na+ transport across cultured AECs is enhanced by exposure to catecholamines and agents that are selective for β2ARs (Kim et al. 1991; Minakata et al. 1998; Bertorello et al. 1999). In experimental lung models, increased Na+ transport after stimulation of β2ARs has resulted in enhanced clearance of pulmonary edema fluid (Saldias et al. 1999,2000; Frank et al. 2000; Azzam et al. 2001). Several mechanisms have been proposed to account for these effects of β2-adrenergic agonists, including an increase in phosphorylation of the Na+,K+-ATPase α-subunits and quantity of the subunit at the basolateral membrane, enhanced Na+ channel open probability, and migration of epithelial Na+ channel (ENaC) components to the apical membrane (Crandall and Matthay 2001). Subacutely, increases in Na+,K+-ATPase α-subunit mRNA and protein and αENaC mRNA expression have been described (Minakata et al. 1998). Indirect enhancement of Na+ absorption after stimulation of β2ARs by promoting apical chloride conductance has also been suggested (Jiang et al. 2001). AT1 cells, by virtue of the fact that their long cytoplasmic processes cover most of the gas exchange surface of the lung, are likely candidates to respond to β2-adrenergic agonists by stimulating Na+ and water clearance. This study demonstrates that AT1 cells possess the receptor for β2-adrenergic agonists.
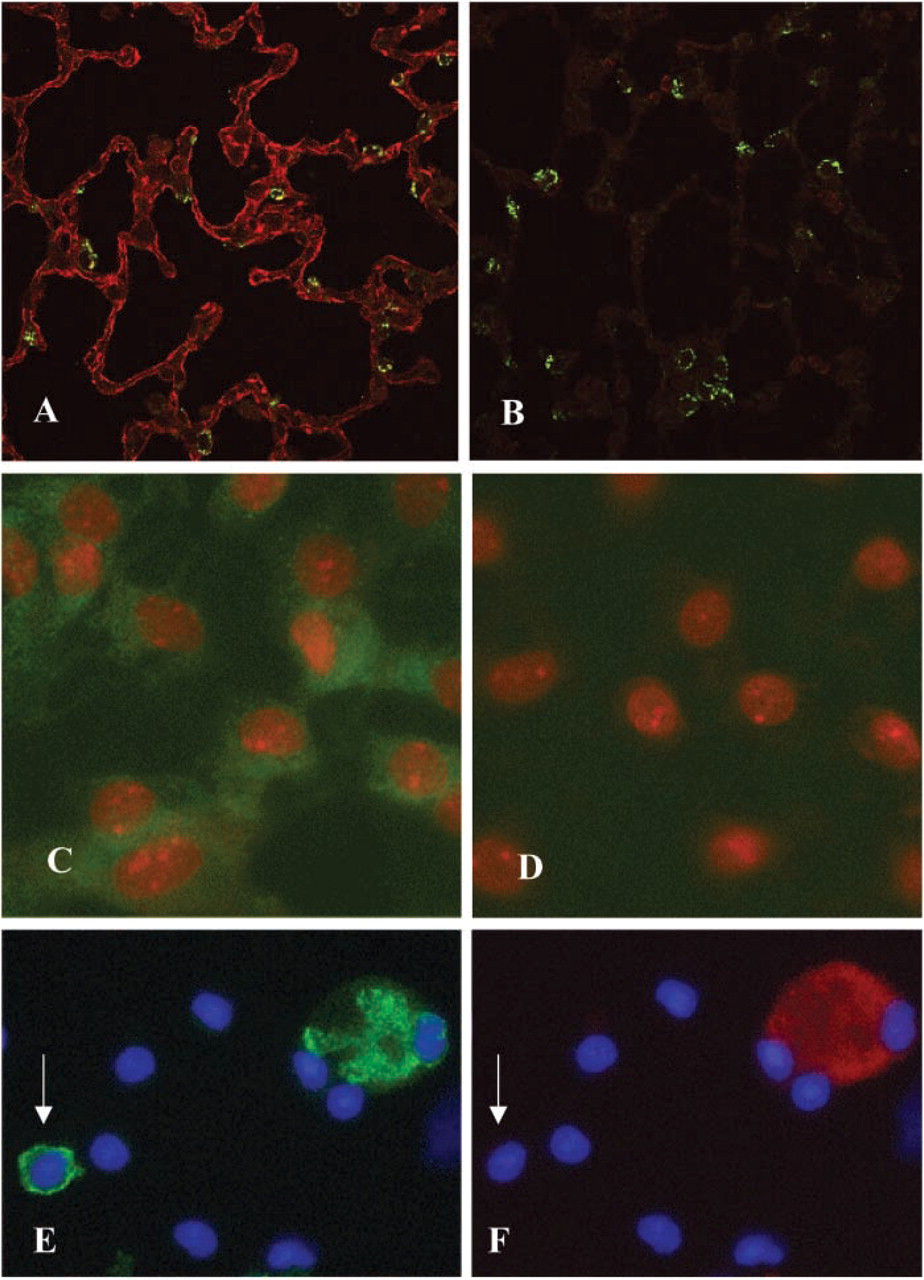
Immunolocalization of GRK2 in whole lung sections of rat lung were double labeled with rabbit anti-GRK2 Ab (red) and goat anti-SP-C Ab (green), a marker for AT2 cells, and viewed using confocal microscopy. (
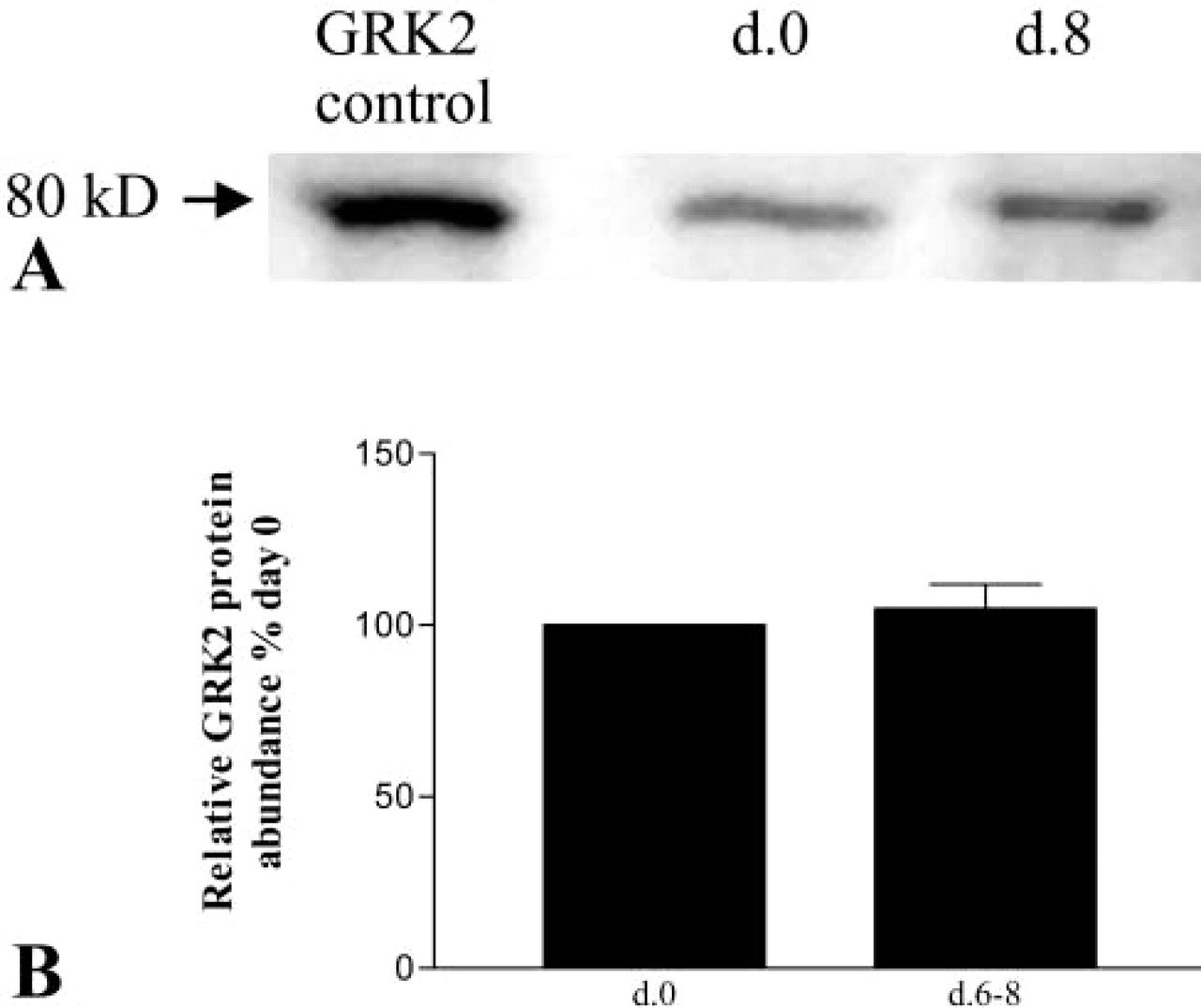
Expression of GRK2 protein in AECs. Western analysis was performed with anti-GRK2 Ab using lysates of either purified freshly isolated AT2 cells (d.0) or AEC in culture that have transdifferentiated to the AT1 cell phenotype (days 6–8). (
Previous attempts to localize β2AR in lung used radioligand receptor labeling or in situ hybridization (Carstairs et al. 1985; Hamid et al. 1991; Mak et al. 1996). Those studies localized β2AR activity and mRNA to alveolar walls but were unable to precisely localize it to AT1 vs AT2 cells. Only recently have satisfactory Abs for immunolocalization of β2AR become readily available (Boivin et al. 2001) that have been used to localize β2AR in other types of cells. In rat kidney, β2AR was localized to apical and subapical areas of proximal and distal renal tubule epithelial cells, with only faint immunoreactivity seen on basolateral membranes (Boivin et al. 2001). Furthermore, localization of β2AR was found to correspond to radioligand binding, indicating that the receptor had intact β2AR activity. In human lung fibroblasts and vascular smooth muscle cells, strong β2AR reactivity was found primarily at the plasma membrane but also occurred in the cytoplasm (Ruiz-Gomez and Mayor 1997). It was speculated that the non-membrane-associated β2AR might reflect cytoplasmic recirculation of the receptor (Ruiz-Gomez and Mayor 1997). Consistent with these previous studies, we found that β2AR immunoreactivity was most intense on the AEC membranes but was also scattered throughout the cytoplasm of the cells.
β2AR signaling occurs via agonist binding to the receptor, which then interacts with the membrane-bound G-protein (Liggett 1997). One feature of G-protein-coupled receptors is the ability to modulate function in response to differing conditions (Liggett 1997). Phosphorylation of G-protein-coupled receptors in general and of β2ARs in particular constitutes a major mechanism for desensitization of these receptors. GRKs are a family of at least six members that differ in tissue distribution, substrate interactions, and regulation (Liggett 1997). The most important GRK that mediates desensitization of β2AR is believed to be GRK2 (McGraw and Liggett 1997). McGraw and Liggett (1997) found significant lung cell-type variation in expression of GRK2 in cultured mast cells, bronchial epithelial cells, and airway smooth muscle cells. They further found a direct relationship between GRK2 expression and the degree of short-term desensitization of these cells to continued agonist exposure. Therefore, even within a single organ such as the lung, cells may experience different degrees of desensitization to β2-adrenergic agents. In this study we demonstrate similar expression of GRK2 in freshly isolated AT2 cells and in AT1-like cells in culture, suggesting that β2AR in both cell types may be influenced by GRK2 in a similar fashion.
Previous investigators have looked for evidence of desensitization of the response to β2-adrenergic agents in vivo and in vitro (Nishikawa et al. 1994; Charron et al. 1999; Kelsen et al. 2000; Morgan et al. 2002). Studies of prolonged exposure to β-adrenergic agonists in rats showed that enhanced clearance of alveolar fluid persisted over 4 hr (Charron et al. 1999), while 48 hr of exposure resulted in a decline in the ability of a β2-adrenergic agonist to stimulate alveolar fluid clearance (Morgan et al. 2002). After 7 days of an inhaled β2-adrenergic agonist, airway epithelial cells harvested from human subjects showed desensitization to this agonist, with an increase in immunoreactivity to GRK2 (Kelsen et al. 2000). These studies suggest that the relative responsiveness of β2ARs of AT1 or AT2 cell origin may diminish over time and may be mediated, at least in part, by GRK2.
In summary, β2AR was immunolocalized to AT1 and AT2 cells in normal rat lung. We further found mRNA for β2AR in AECs in primary culture that had undergone transdifferentiation towards an AT1 cell-like phenotype. These findings suggest a role for AT1 cells in the increase in alveolar Na+ and fluid clearance observed after exposure to catecholamines or more selective β2- adrenergic agents. Both AT1 and AT2 cells in whole lung express immunoreactive GRK2. The effect of GRK2 on relative responsiveness of different cells in alveolar epithelium to continued β2-adrenergic agonists awaits further study. We speculate that the net effect of β2-adrenergic agonists on clearance of pulmonary edema fluid will depend on the balance among the numbers of β2ARs available on each cell type, the numbers of each cell type present, and the relative impact of β2AR regulators, such as GRK2, on each cell type. Unknown at this point is the degree to which this dynamic interplay of cells-receptors-desensitizers occurs in the setting of lung injury, when the usual relationship between AT1 and AT2 cells is disrupted and hyperplastic AT2 cells may be the predominant type of epithelial cell present. Characterization of these features in AT1 and AT2 cells under normal conditions and in lung injury will lead to a better understanding of the potential role of β2-adrenergic agonists in the management of pulmonary edema.
Footnotes
Acknowledgements
Supported by National Heart, Lung and Blood Institute Research Grants HL-03609, HL-38578, HL-38621, HL-38658, HL-51928, HL-62569, and HL-64365, and by the Hastings Foundation.
We note with appreciation the excellent technical support of Zerlinde Balverde and Susie Parra. The Microscopy Sub core at the USC Center for Liver Diseases (NIH 1 P30 DK48522) provided the confocal microscope used for these studies. Dr Crandall is Hastings Professor and Norris Chair of Medicine.