
Abstract
The temporal and spatial expression of transforming growth factor (TGF)-β1 and connective tissue growth factor (CTGF) was assessed in the left ventricle of a myocardial infarction (MI) model of injury with and without angiotensin-converting enzyme (ACE) inhibition. Coronary artery ligated rats were killed 1, 3, 7, 28, and 180 days after MI. TGF-β1, CTGF, and procollagen α1(I) mRNA were localized by in situ hybridization, and TGF-β1 and CTGF protein levels by immunohistochemistry. Collagen protein was measured using picrosirius red staining. In a separate group, rats were treated for 6 months with an ACE inhibitor. There were temporal and regional differences in the expression of TGF-β1, CTGF, and collagen after MI. Procollagen α1(I) mRNA expression increased in the border zone and scar peaking 1 week after MI, whereas collagen protein increased in all areas of the heart over the 180 days. Expression of TGF-β1 mRNA and protein showed major increases in the border zone and scar peaking 1 week after MI. The major increases in CTGF mRNA and protein occurred in the viable myocardium at 180 days after MI. Long-term ACE inhibition reduced left ventricular mass and decreased fibrosis in the viable myocardium, but had no effect on cardiac TGF-β1 or CTGF. TGF-β1 is involved in the initial, acute phase of inflammation and repair after MI, whereas CTGF is involved in the ongoing fibrosis of the heart. The antifibrotic benefits of captopril are not mediated through a reduction in CTGF.
Several studies have assessed the role of cytokines on myocyte injury and myocardial function in the acute phase after MI (Ohnishi et al. 1998; Ono et al. 1998; Sun et al. 1998; Lijnen et al. 2000; Yu et al. 2001; Ahmed et al. 2004). Low levels of expression of transforming growth factor (TGF)-β1, its receptor, and collagen type 1 are seen in normal rat heart and increase after MI. The increase in TGF-β1 mRNA precedes increases in collagen expression, suggesting a role for TGF-β1 in cardiac fibrosis and remodelling (Youn et al. 1999; Yu et al. 2001; Tzanidis et al. 2001). Connective tissue growth factor (CTGF) has also been shown to promote fibroblast proliferation and extracellular matrix production in connective tissues, and its overexpression has been observed in wound repair and in fibrotic disorders (Igarashi et al. 1993). Acutely after MI, the expression of CTGF increases in parallel with collagen (Ohnishi et al. 1998).
The concomitant regional and spatial changes in expression of TGF-β1 and CTGF after MI have not been assessed. Many studies have assessed either TGF-β1 or CTGF and most have been short term (<6 weeks) in nature (Ohnishi et al. 1998; Youn et al. 1999; Tzanidis et al. 2001; Yu et al. 2001; Ahmed et al. 2004). To date, no studies have assessed the expression of TGF-β1 or CTGF in the chronic stage of remodelling when heart failure has occurred. We have previously shown that significant changes in the spatial distribution of other cytokine systems such as the insulin-like growth factor system can occur for up to 6 months after infarction when heart failure is present (Dean et al. 1999). It is possible that sustained production of profibrotic cytokines may underlie the development of fibrosis in the remodelling and failing heart.
We have previously described the benefits of angiotensin-converting enzyme (ACE) inhibition to reduce both cardiac remodelling and fibrosis after MI in the rat (Burrell et al. 2000), and now hypothesize that the long-term beneficial effect of ACE inhibitors may be mediated through changes in the profibrotic cytokines TGF-β1 or CTGF. The aims of this study were 2-fold: (1) to examine the time course and cellular expression of TGF-β1, CTGF, and collagen in the heart for up to 180 days after MI when heart failure is present and (2) to assess whether one of the mechanisms by which an ACE inhibitor attenuates cardiac remodelling and fibrosis is modulation of either or both of these profibrotic cytokines.
Materials and Methods
Experimental procedures were performed according to the National Health and Medical Research Council of Australia Guidelines for Animal Experimentation. Rats were housed at 23 to 25C in a 12:12 light:dark cycle, with ad libitum food containing 0.4–0.6% NaCl and water.
Experimental Design
The rat model of MI has been extensively used to examine the efficacy of therapeutic interventions on cardiac remodelling and survival (Pfeffer et al. 1985; Burrell et al. 1996, 2000; Dean et al. 1999). Left ventricular free-wall myocardial infarction was induced in female Sprague-Dawley rats (200–250 g) by ligation of the proximal left anterior descending artery as described previously. Sham-operated (control) rats underwent an identical operation, but the suture was not tied.
Study 1: Time Course
At 1, 3, 7, 28, and 180 days after MI, rats (
Study 2: Captopril Treatment
In a separate study, rats surviving for 24 hr postoperatively were randomized to vehicle (once daily gavage of 5% arabic gum) or captopril (25 mg/kg, twice daily gavage) (
Infarct Size
The LV was sectioned at four levels from the base to the apex, paraffin embedded, and sections cut and stained with Masson's trichome, and hematoxylin and eosin. The mean epicardial and endocardial scar circumference was compared with total LV circumference to calculate total infarct size (Pfeffer et al. 1985).
In Situ Hybridization
In situ hybridization was performed on 4-μm LV sections from rats involved in study 1 and 2 (
Antisense and sense riboprobes labeled with 35S cytidine 5'-T trisphosphate (CTP) were prepared using the Promega transcription system. Between 500 ng and 1 μg of linearized template cDNA was transcribed in a reaction mix containing 1 X transcription buffer; 10 mM DTT; 0.6 mM each of ATP, GTP, and UTP; 100 μCi [35S]; 0.5 μl RNasin; and 1 μl of the appropriate RNA polymerase. If the probe length was greater than 400 bp, 4.25 pM of cold CTP was added to the transcription reaction. The reaction was incubated at 37C for 90 min; after this time, 1000 U of DNase (RNase-free) (Boehringer Mannheim; Roche Diagnostics, Sydney, Australia) was added and the reactions incubated for a further 15 min at 37C. The riboprobe was precipitated with ammonium acetate and ethanol using yeast tRNA as carrier, then reconstituted in 10 mM DTT. The length of the purified riboprobe was adjusted to ≃150 bp using alkaline hydrolysis, followed by further purification with sodium acetate and ethanol, and resuspension in 10 mM DTT.
Heart sections were dewaxed, rehydrated, digested with Pronase E at 37C, and hybridized overnight at 60C with a buffer containing 2 X 104 cpm/μl of 35S-labeled riboprobe, 0.72 mg/ml yeast RNA, 50% deionized formamide, 100 mM DTT, 10% dextran sulfate, 0.3 M NaCl, 10 mM Na2HPO4, 10 mM Tris-HCl (pH 7.5), 5 mM EDTA (pH 8.0), 0.02% BSA, 0.02% Ficoll 400, and 0.02% polyvinyl pyrrolidone.
After hybridization, sections were stringently washed with 50% formamide, 2 X SSC at 55C, incubated with RNase A (150 μg/ml), dehydrated and exposed to Kodak X-Omat autoradiographic film (Eastman Kodak, Rochester, New York) for 1–3 days at room temperature. Slides were dipped in photographic emulsion (Amersham; Buckinghamshire, UK), stored with desiccant at 4C for 14–21 days, developed in Kodak D19, fixed in Ilford Hypam, and stained with hematoxylin and eosin for cellular localization.
Quantitation of Macroscopic In Situ Hybridization Autoradiographs
Quantitation was carried out using a microcomputer imaging device (Imaging Research, Ontario, Canada) run by an IBM PC. Sections from four levels of the LV from each animal were used for quantitation. In MI hearts, the viable myocardium, scar, and border zones were quantitated separately. The border zone is the area of high cellular infiltrate at the edge of the fibrotic scar tissue of the infarct. The optical densities of the autoradiographs were calibrated in terms of radioactivity density as dpm/mm2 by reference to radioactive standards (Amersham, UK) carried through the procedures (Le Moine et al. 1994). Quantitation was carried out in the linear region of the curve generated by the radioactive standard. Results are expressed as specific labeling (using antisense probes) minus nonspecific labeling (using sense probes). Nonspecific labeling with the antisense probe was also checked by the use of “negative tissues” for each probe.
Total Collagen Staining
Paraffin sections 4 μm thick were deparaffinized, rehydrated, and then stained with 0.1% Sirius Red (Polysciences Inc.; Warrington, PA) in saturated picric acid (picrosirius red) for 1 hr, differentiated in 0.01% HCl for 30 sec, and rapidly dehydrated. Collagen volume fraction was determined by measuring the area of stained tissue with in a given field. Within the LV, fields containing vessels, artifacts, minor scars, or incomplete tissue were excluded. A total of 15–20 fields were analyzed per animal. The area stained was calculated as a percentage of the total area within a field (Yu et al. 1998). Total collagen volume fraction determined by this morphometric approach is closely related to the hydroxyproline concentration in the LV (Weber et al. 1988).
Immunohistochemistry for ED1, TGF-β1, and CTGF
Immunohistochemistry for macrophages and TGF-β1 was carried out on 4-μm paraffin sections of LV as previously described (Gilbert et al. 1998; Dean et al. 1999). For CTGF, the primary antibody used was a polyclonal rabbit antimouse CTGF antibody (Abcam Ltd, Cambridge, UK) at a concentration of 1:800. Standard techniques were employed and the Elite Vectastain ABC kit (Vector Laboratories, Burlingame, CA) was used. The staining was visualized by reaction with 3,3'-diaminobenzidine tetrahydrochloride (Sigma Chemical Co., St Louis, MO). Negative control sections were incubated in the absence of primary antibody.
Immunohistochemical staining for TGF-β1 and CTGF protein was quantitated (
Statistics
Results are expressed as mean ± SEM. Comparisons of tissue weights and vasoactive hormone levels were made by unpaired
Results
Study 1: Time Course
Silver grains representing procollagen α1(I) mRNA were seen overlying inflammatory cells throughout the myocardium and surrounding vessels of the scar and border zones of the infarcted LV and to fibroblasts surrounding vessels in all areas of the infarcted LV (not shown). In addition, cords of connective tissue throughout the non-infarcted myocardium demonstrated procollagen α1(I) labeling (not shown).
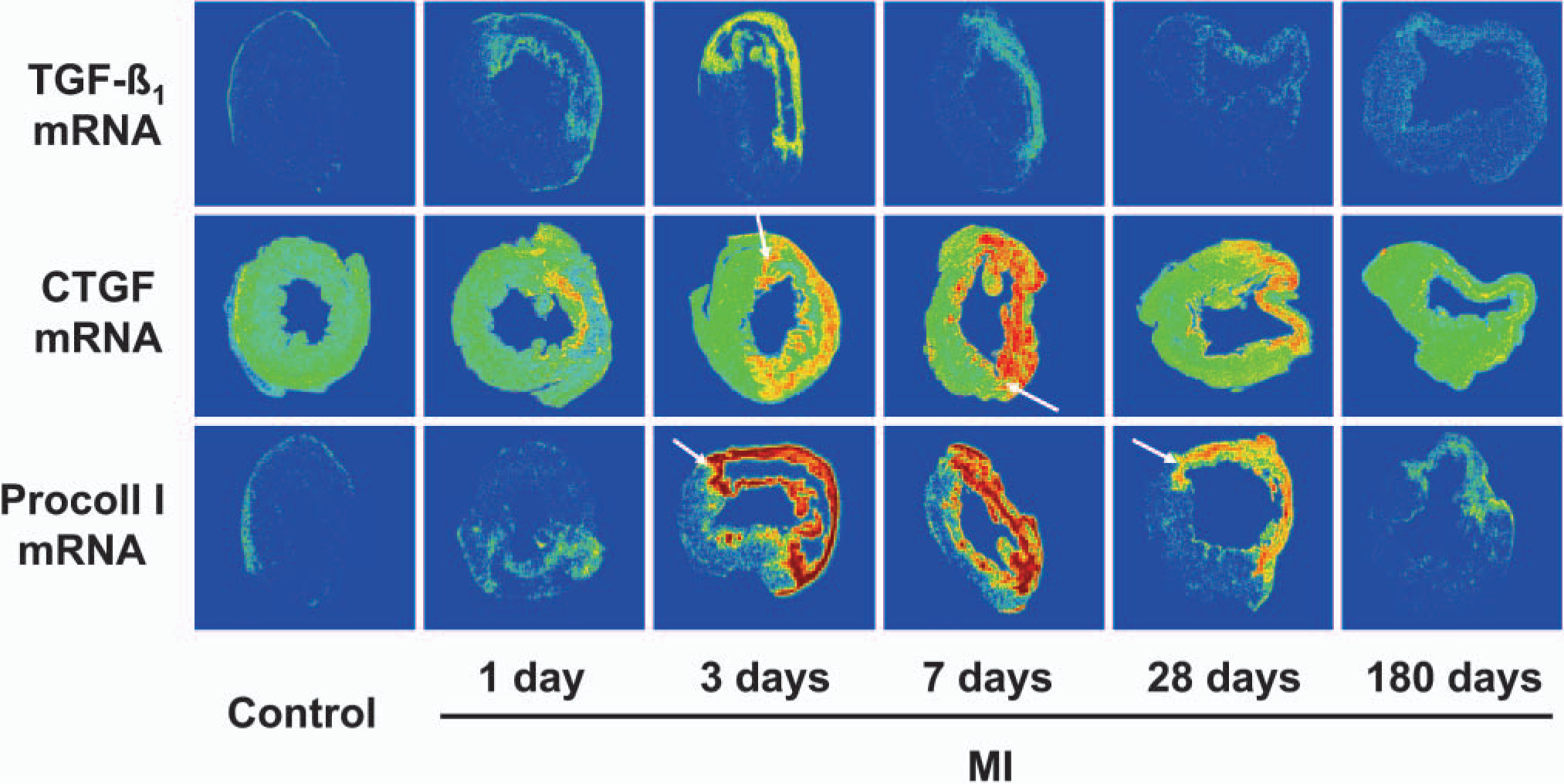
Representative macroscopic autoradiographs demonstrating levels of signal produced by in situ hybridization histochemistry for transforming growth factor-β1 (top row), connective tissue growth factor (middle row), and procollagen α1(I) (bottom row). Increasing levels of signal are demonstrated by a color scale, with red being the highest levels of radiolabeling and blue the lowest. Comparisons can only be made between time points and not individual mRNA species. The scar of the myocardial infarction (MI) left ventricle is present where thinning of the left ventricular wall has occurred. The scar is surrounded by the border zone seen as a ring of high density labeling at 3 days after MI and by 28 days as the junction between the thinned wall and the remaining viable myocardium (white arrows).
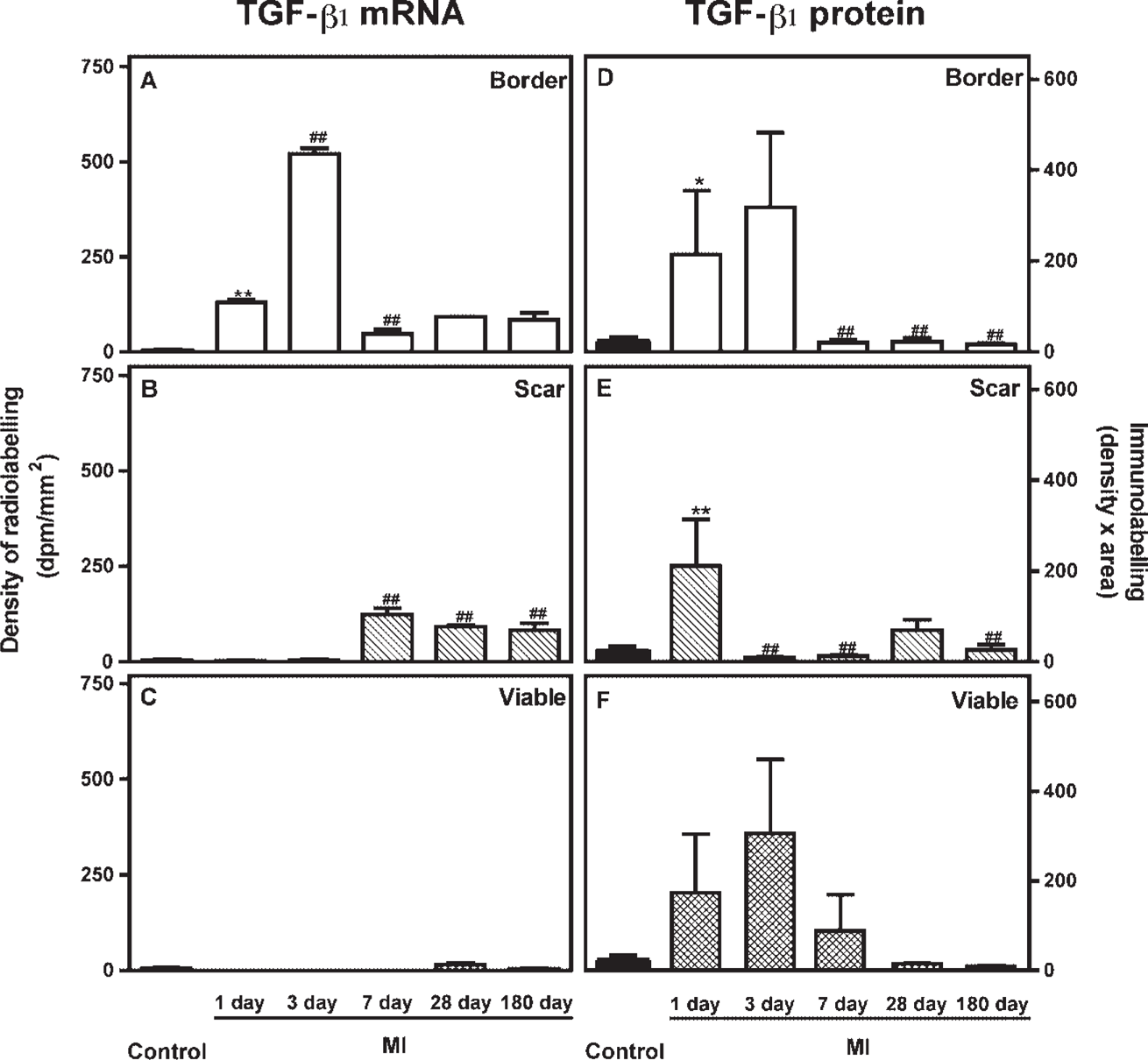
TGF-β1 mRNA localized to the same areas as procollagen α1(I) mRNA. Immunohistochemical analyses showed the major cell population to be macrophages, the level of labeling tended to decrease over the time course and was at very low levels by 180 days, as shown previously (Dean et al. 1999). TGF-β1 mRNA labeling was low in control LV.
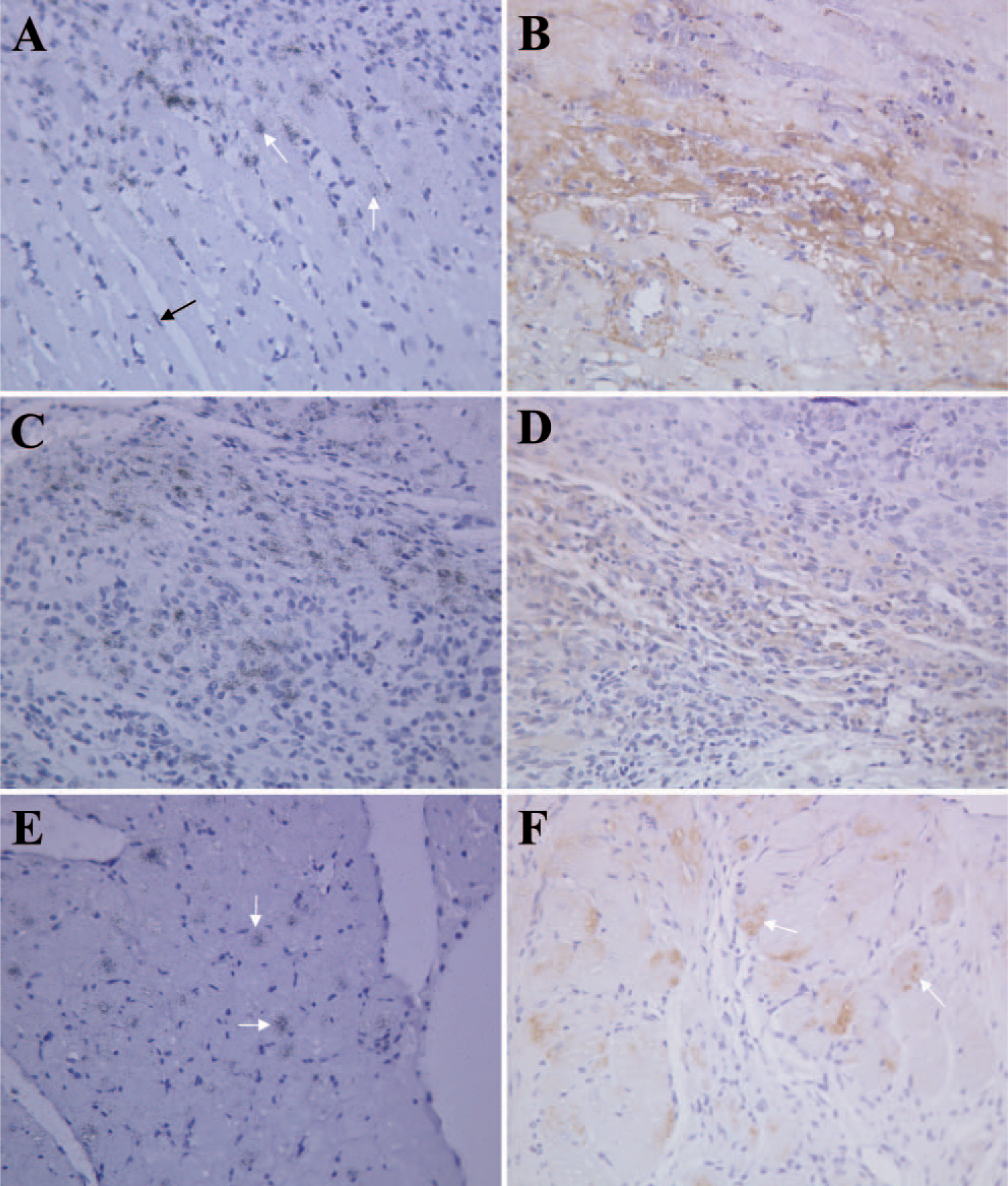
Micrographs of connective tissue growth factor (CTGF) in situ hybridization
Expression of procollagen α1(I) mRNA
Study 2: Captopril Treatment
All sham-operated rats (
Expression of transforming growth factor-β1 mRNA
Discussion
The main findings of this study in rat after MI are that (1) temporal and regional differences in the expression of TGF-β1, CTGF, and collagen occur; (2) by 180 days after MI, significant fibrosis is present in the viable myocardium and is also associated with the profibrotic cytokine CTGF but not TGF-β1; and (3) treatment with an ACE inhibitor attenuates cardiac remodelling, reduces LV mass, and decreases fibrosis in the viable myocardium, but these effects are not mediated by changes in cardiac CTGF expression.
Fibrosis and Cytokine Expression after MI
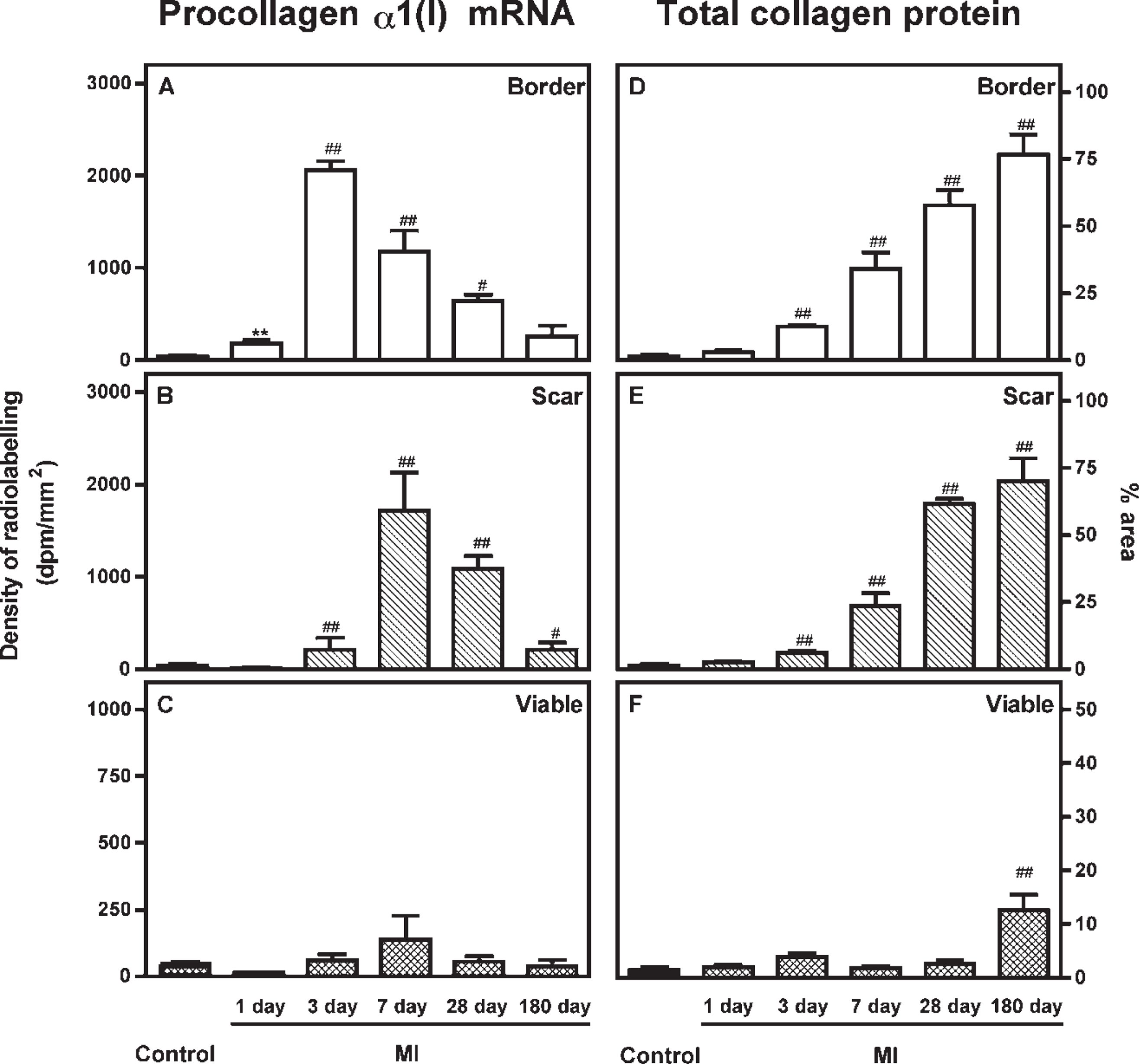
It has been suggested that TGF-β1 or CTGF overexpression is involved in cardiac fibrosis and that strategies to reduce these cytokines may be useful in heart failure. In this study, low-level expression of TGF-β1, CTGF, and collagen mRNA were seen in normal rat hearts. TGF-β1 increased acutely after MI, preceding increases in collagen expression. The finding that TGF-β1 and procollagen α1(I) mRNA expression were localized to infiltrating inflammatory cells and fibroblasts of the scar and border zones early after MI and were not increased in the distant non-infarcted myocardium suggests a regulated response to injury and mechanical stress. It is, however, feasible that localization of procollagen α1(I) mRNA in inflammatory cells is a nonspecific result and does not result in translation to protein as in fibroblasts. These results extend observations of previous studies that have shown at 2 days after MI there is increased expression of TGF-β1 mRNA and protein in the border zone (Thompson et al. 1988) and at 1 week after MI, TGF-β1 mRNA is induced in non-myocytes (Yue et al. 1998). By 8 weeks after MI, Western blot analysis of cardiac tissue showed the active form of TGF-β1 was significantly elevated in border and scar tissue, but myocytes remote to the infarct zone expressed comparatively moderate levels of TGF-β1 only (Hao et al. 2000). We found parallel increases in TGF-β1 mRNA and protein in the early stages after MI. With time, however, the levels of protein fell to control values in all areas of the heart. Interestingly, in the scar and border zone, TGF-β1 mRNA remained high, but was not translated to protein.
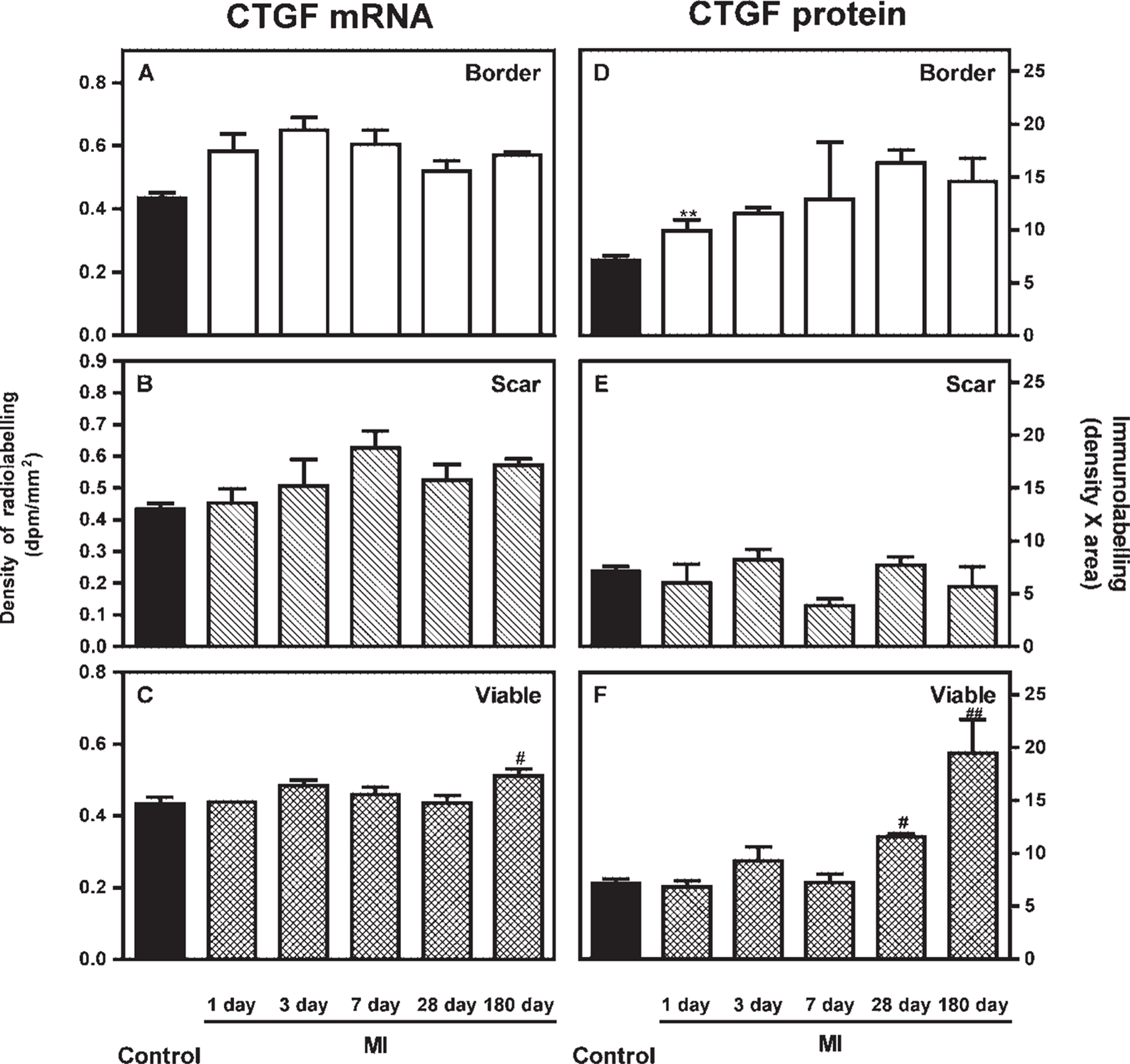
Expression of connective tissue growth factor mRNA
In contrast, CTGF mRNA and protein were increased in myocytes and non-myocytes of the non-infarcted myocardium 180 days after MI, an increase associated with fibrosis. This extends previous reports that demonstrate that CTGF is increased in non-myocytes 42 days after MI (Ahmed et al. 2004).
Our data suggest that TGF-β1 may be involved in the initial induction of procollagen α1(I) mRNA and total collagen in the early stages after MI, but is not important in the laying down of collagen protein in the continuing remodelling of the heart. The localization of TGF-β1 mRNA to inflammatory cells suggests that TGF-β1 has a role beyond collagen production, possibly pleiotropic actions such as wound healing or simply maintaining physiological functioning of the heart (Azhar et al. 2003). On the other hand CTGF is involved in remodelling of the viable myocardium in the chronic stage after MI, when the inflammatory response has subsided, but fibrosis is ongoing.
ACE Inhibition after MI
ACE inhibitors are standard therapy after myocardial infarction to attenuate remodelling, delay the progression to heart failure, and decrease mortality (Pfeffer et al. 1992). Despite the benefits of ACE inhibitors to slow remodelling, progressive increases in LV dilation and hypertrophy continue to occur, in part because of continued cytokine activation and ongoing fibrosis (Eichorn and Bristow 1996). Although it is clear that fibrosis of the scar is an important and necessary response to injury, fibrosis remote to the scar, in the viable non-infarcted myocardium, contributes adversely to tissue structure and function and to the development of heart failure (Weber 1997).
Renin-angiotensin System Blockade and TGF-β1
Both in vitro and in vivo studies have shown Ang II stimulates TGF-β1 synthesis (Dostal et al. 1996; Border and Noble 1998). It is well known that fibrosis and activation of the cardiac renin-angiotensin system (RAS) occurs after MI (Passier et al. 1996; Duncan et al. 1997), and we have shown that the ACE inhibitor captopril reduces LV mass after MI (Burrell et al. 2000) and has antifibrotic effects in the heart (Farina et al. 2000). Although these data suggest a link between the RAS, fibrosis and the TGF-β1 system, studies with ACE inhibitors or Ang II type 1 receptor antagonists have produced conflicting results in terms of whether RAS blockade does (Sun et al. 1998; Youn et al. 1999; Yu et al. 2001) or does not (Tzanidis et al. 2001; Yu et al. 2001) “switch off” cardiac TGF-β1 expression after MI. In this study, 180 days of treatment with captopril had no effect on TGF-β1 levels, which may be explained by the fact that the TGF-β1 levels had already returned to base levels at this time. However, as expected, ACE inhibition did decrease collagen protein in the viable myocardium of MI rats.
Infarct size, blood pressure, tissue weights, and blood hormones
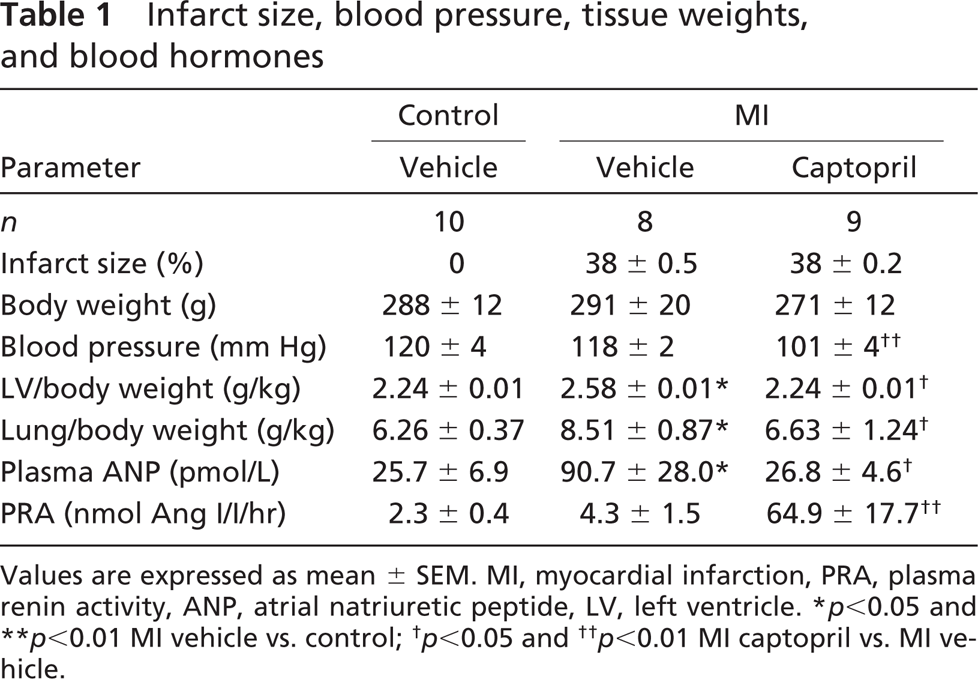
Values are expressed as mean ± SEM. MI, myocardial infarction, PRA, plasma renin activity, ANP, atrial natriuretic peptide, LV, left ventricle. ∗
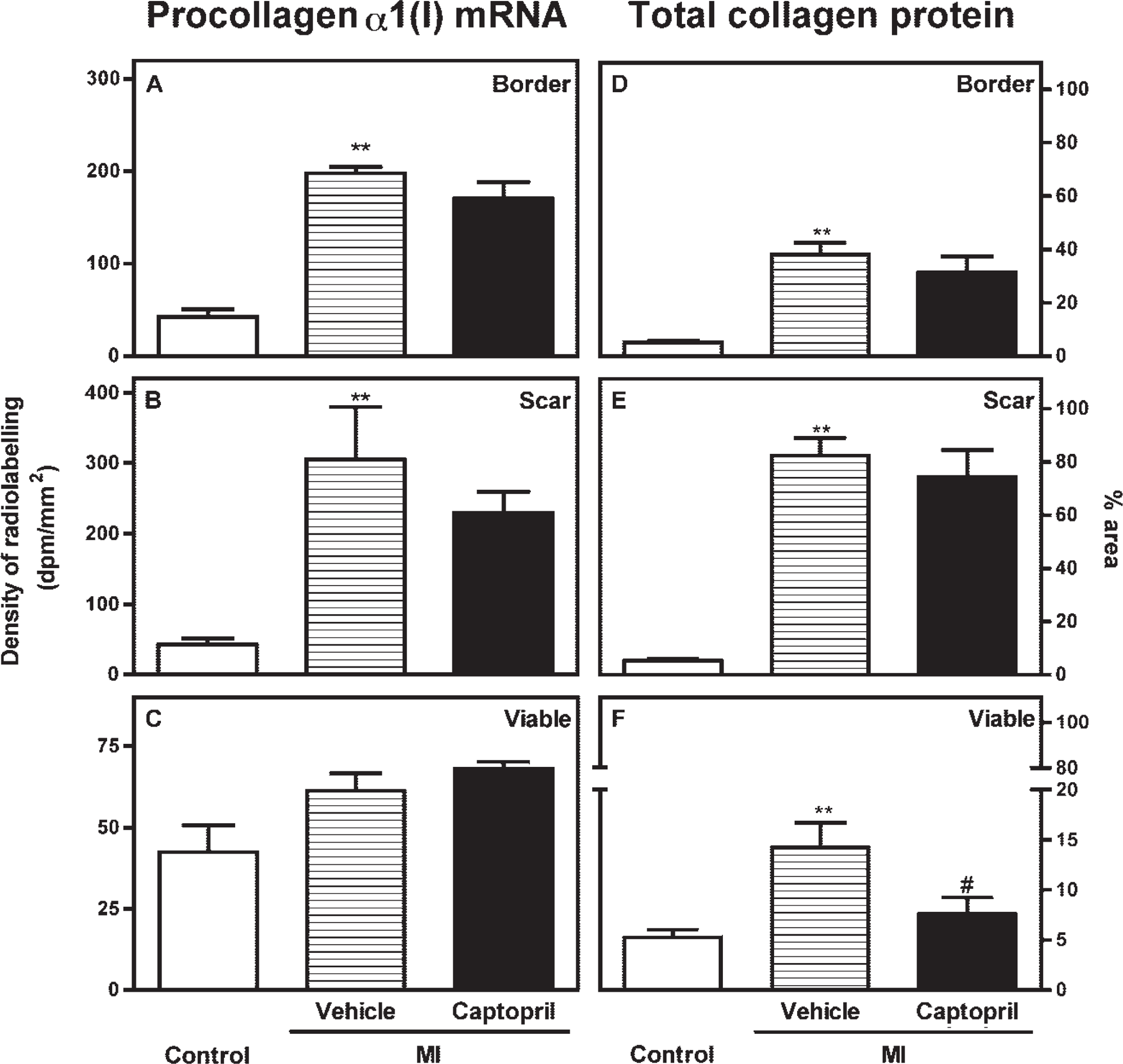
Effect of captopril treatment on the expression of procollagen α1(I) mRNA
RAS Blockade and CTGF
Data on RAS blockade and its effect on CTGF expression are limited. In this study, chronic activation of left ventricular CTGF mRNA and protein was not prevented by ACE inhibition. However, in vascular smooth muscle cells, an Ang II type 1 receptor antagonist blocked Ang II-induced CTGF gene and protein expression (Ruperez et al. 2003). In a similar model to that used in our study, the AT1 receptor antagonist losartan prevented the induction of myocardial CTGF mRNA after 25 days of treatment; however, its effect on CTGF protein was not examined (Ahmed et al. 2004). This suggests that there may be a difference in treatment outcome between ACE inhibition and angiotensin receptor blockade or that the treatment effect has “worn off” after 6 months of treatment.
TGF-β1 Induction of CTGF
It was originally believed that the main mechanism for upregulation of CTGF expression was induction by TGF-β1 in both cardiac (Chen et al. 2000) and non-cardiac tissues (Yokoi et al. 2001). Data from this study do not support this mechanism because the levels of TGF-β1 mRNA have returned to baseline levels well before the activation of CTGF. It is now becoming clear that there are alternative mechanisms to induce CTGF expression including activation of a G protein-coupled receptor (Hahn et al. 2000), blood pressure-dependent mechanisms (Finkenberg et al. 2003), and protein kinase C activation (Way et al. 2002). Way et al. demonstrated that, in the fibrotic myocardium of transgenic mice overexpressing protein kinase C, dilating, and undergoing hypertrophy, CTGF, but not TGF-β expression was increased at all time points studied. Furthermore, consistent with our earlier studies of advanced glycation effects in mesenchymal cells (Twigg et al. 2001) in the diabetic rat heart, CTGF, but not TGF-β expression is increased (Way et al. 2002; Candido et al. 2003). Alternatively, CTGF has been shown to potentiate TGF-β1, by facilitating TGF-β1 binding to the TGF-β receptor and that, although in this study TGF- β1 itself does not rise at 180 days, it may well be that TGF-β1 signaling pathways are used and potentiated by the increase in CTGF protein (Abreu et al. 2002).
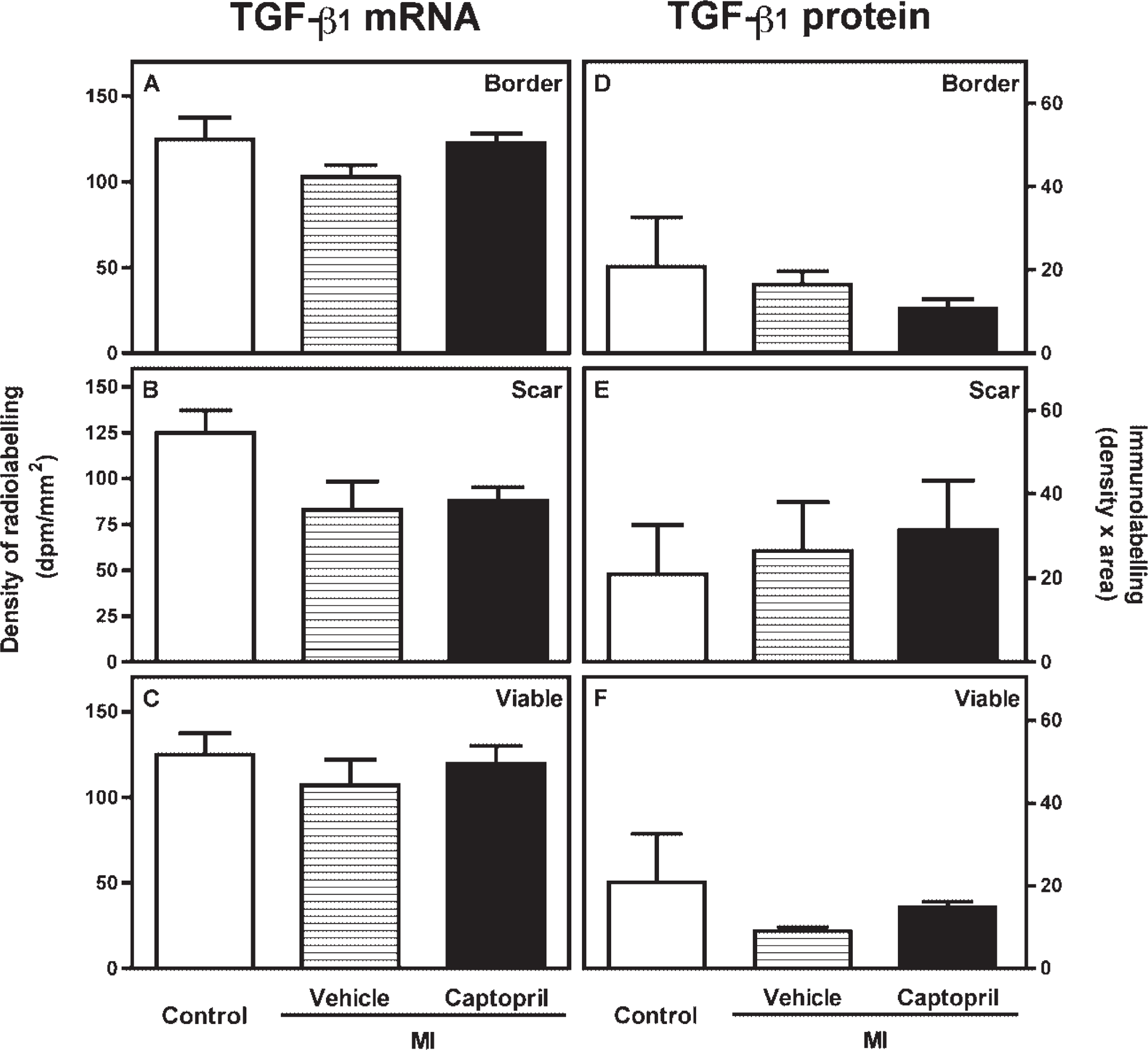
Effect of captopril treatment on the expression of transforming growth factor-β1 mRNA
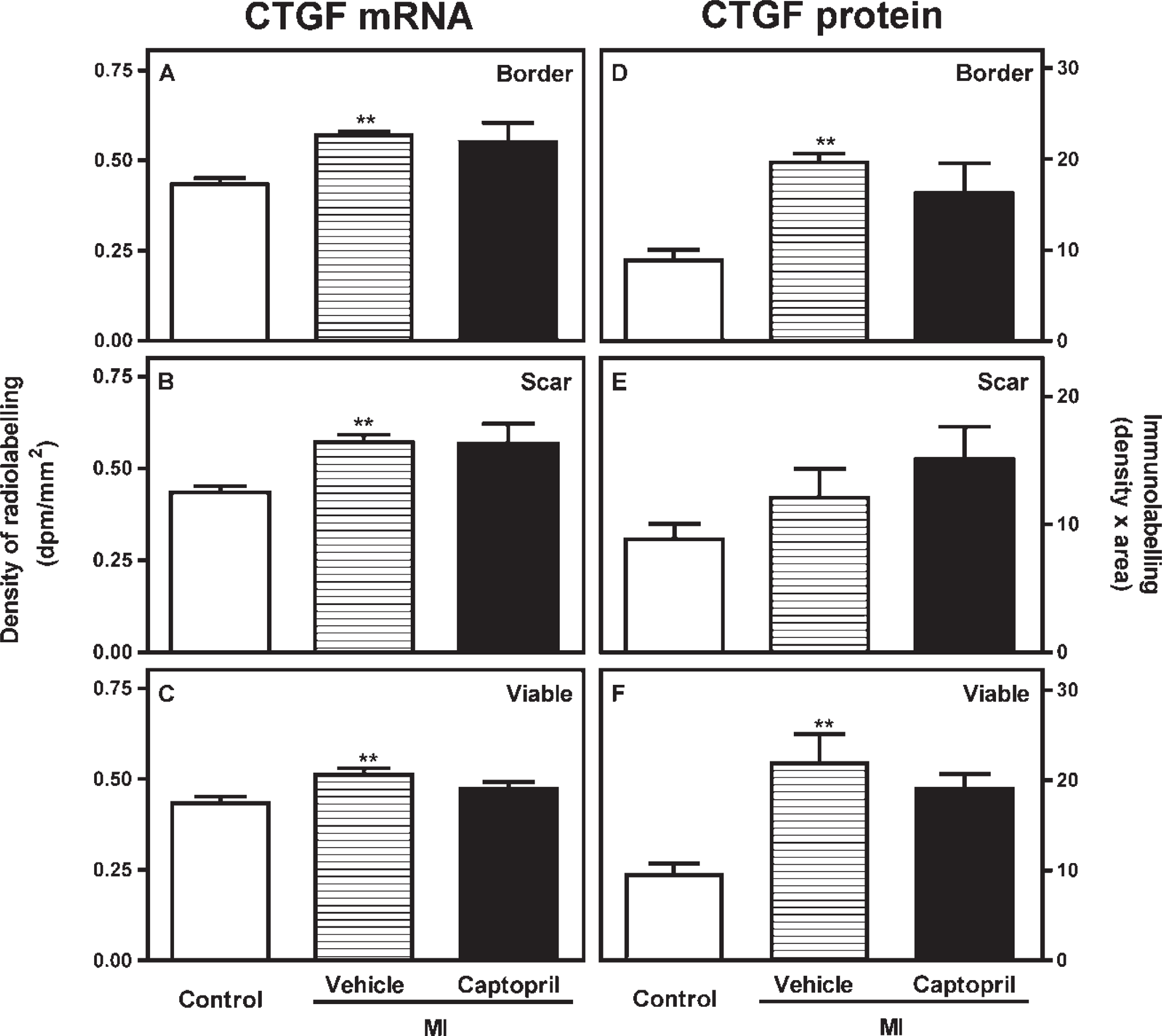
Effect of captopril treatment on the expression of CTGF mRNA
Limitations
It is possible that small non-significant changes in cytokine levels may lead to significant effects on tissue remodelling and structure. Future in vitro studies may give some insight as to the magnitude of change in CTGF required to modulate collagen production and thus fibrosis, as well as to assess the effect of RAS blockade on this process.
Summary
This study provides evidence that TGF-β1 plays a role in the acute response to myocardial injury and that CTGF is involved in the late phase of post-MI remodelling when injury and inflammation have abated, but mechanical load remains high and the non-infarcted myocardium is fibrosing, dilating, and hypertrophying. Furthermore, the cardiac benefits of captopril to reduce fibrosis in the viable myocardium are not mediated through changes in CTGF. Identification of a therapeutic intervention that reduces CTGF in the late phase of post-MI remodelling may prove beneficial in the search for improved treatment regimes in cardiovascular disease.
Footnotes
Acknowledgements
This study was supported by the Sir Edward Dunlop Medical Research Foundation.
The authors wish to acknowledge the assistance of Dr. Richard E. Gilbert, Mr. Anthony Gleeson, Mrs. Donna Paxton, and Mrs. Alison Cox.