
Abstract
Hypertension is a common complication in children with autosomal recessive polycystic kidney disease (ARPKD) who have survived the neonatal period. No information is available regarding the mechanism of hypertension in this condition. The renin-angiotensin system (RAS) is thought to play a role in hypertension associated with the more common autosomal dominant polycystic kidney disease (ADPKD). Occasional reports have documented increased activity of the intrarenal RAS in ADPKD, with ectopic renin expression within cysts and dilated tubules. Because of similarities between ARPKD and ADPKD, we hypothesized that increased intrarenal RAS activity might also be found in ARPKD. We performed immunohistochemical studies on kidney tissues from two infants with ARPKD and two control kidneys. The cystic dilated tubules showed staining with the peanut lectin arachis hypogaea, a marker of distal tubules and collecting ducts, but not with lotus tetragonolobus, a marker of proximal tubules. Strong renin staining was seen in many cysts and tubules of ARPKD kidneys, but only in the afferent arterioles of the normal control kidneys. Angiotensinogen staining was also observed in some cysts and in proximal tubules. Staining for angiotensin-converting enzyme, angiotensin II type 1 receptor, and angiotensin II peptide was present in many cystic dilated tubules. These immunohistochemical studies document for the first time ectopic expression of components of the RAS in cystic-dilated tubules of ARPKD and suggest that overactivity of RAS could result in increased intrarenal angiotensin II production, which may contribute to the development of hypertension in ARPKD.
A
Extracellular volume expansion has been reported in both ADPKD and ARPKD patients before the onset of renal failure (Kaplan et al. 1989; Nash 1977). A few studies have examined the involvement of the RAS in hypertension in ADPKD patients, but no consistent relationship has been found between blood pressure and plasma renin activity (Chapman et al. 1990; Seeman et al. 1997). It is now well accepted that increased activity of the intrarenal rather than the systemic RAS is involved in many forms of hypertension (Davisson et al. 1999; Navar et al. 2002). It is postulated that persistent elevations of intrarenal angiotensin II (Ang II) production coupled with the inability to reduce Ang II formation in response to a high sodium intake, will lead to a resetting of the pressure-natriuresis relationship toward higher blood pressures, thus leading to hypertension (Gross et al. 1994; Hall et al. 1996,1999).
We recently found that renin and other components of the RAS are expressed by cysts and dilated tubules in ADPKD kidneys (Loghman-Adham et al. 2004). Our results confirmed and extended earlier reports by Torres et al. (1992) of renin immunostaining in cystic tubules and cysts of ADPKD kidneys. Despite different genetic basis and different gene products, ARPKD and ADPKD share many phenotypic features such as renal cysts and a high prevalence of hypertension. Because of similarities between ARPKD and ADPKD, we hypothesized that ectopic expression of components of the RAS may also be a feature of ARPKD kidneys. We undertook the present immunohistochemical studies on kidney tissues obtained from two infants with ARPKD who had died in the neonatal period and two infants who had died from causes other than kidney disease. We show the ectopic presence of immunoreactive renin and angiotensinogen (AGT) and other components of the RAS in tubules and cysts of ARPKD kidneys. The results provide further support for the hypothesis that, in polycystic kidney disease, ectopic overexpression of components of the RAS could result in increased intrarenal and intratubular Ang II production. Sustained increases in Ang II concentrations might result in hypertension via resetting of the pressure-natriuresis mechanism.
Materials and Methods
Sources of Kidney Tissue
Formalin-fixed, paraffin-embedded polycystic and control kidney blocks were obtained from pathology department archives at Cardinal Glennon Children's Hospital. The procedures for the use of human kidney specimens were approved by St Louis University Institutional Review Board. The two ARPKD kidney specimens used were from infants who had died in the neonatal period. One infant, born at 36 weeks’ gestation, had severe respiratory distress secondary to pulmonary hypoplasia and bilateral pneumothoraces. She required intubation, bilateral chest tubes, and intensive medical care. She remained anuric and eventually died at 10 hr of age. The other infant also died during the neonatal period, but no clinical information is available. The control kidneys used in these studies were from infants who had died from causes not associated with kidney disease. One infant was born at 38 weeks’ gestation and died at 17 days from complications resulting from diaphragmatic hernia and pulmonary dysplasia. The other was a 2-year-old infant who died 3 days after hospital admission from complications of purulent bacterial meningitis.
Immunohistochemistry of Tissue Sections
Four-micron tissue sections were used for immunohistochemistry. The sections were deparaffinized in HemoDe (Fisher Scientific, Pittsburgh, PA), then rehydrated in graded alcohols. Antigen retrieval was performed with 0.01 M citrate buffer, pH 6.0, at 60C for 30 min. The slides were rinsed twice with phosphate-buffered saline (PBS), followed by the addition of 0.6% H2O2 in 20% methanol for 20 min at room temperature to block endogenous peroxidase. They were washed three times with PBS, then blocked with normal horse serum for 20 min at room temperature. The primary antibodies were added at dilutions indicated in Table 1. The slides were incubated either 1 hr at room temperature or overnight at 4C, then washed three times with PBS/0.1% Tween-20, followed by the addition of the second biotinylated antibody and incubated for 30 min at room temperature. For lectin-binding studies, biotinylated lotus tetragonolobus (LTA) and arachis hypogaea (PNA) lectins were used directly at this stage. LTA is a marker of proximal tubules and PNA is a marker of distal and collecting tubules. The slides were washed three times with PBS, followed by the addition of 1 drop of the ABC reagent (Vectastain Elite kit, Vector Laboratories, Burlingame, CA) and incubated at room temperature for 30 min. The slides were washed three times with PBS, followed by the addition of peroxidase substrate for 6–10 min. They were rinsed in distilled water, counterstained with hematoxylin (Gill No 3, Sigma Diagnostics, St Louis, MO) for 60–90 sec, washed extensively in running water, and mounted. They were viewed with a Zeiss Axioplan microscope or with an Olympus CX41 microscope, equipped with a DP12 digital camera, and photographed.
Reagents and Supplies
Rabbit polyclonal anti-human renin antibody (antiserum #74, titer 13,300) used in this study was raised against purified human kidney renin and has been previously described (Celio and Inagami 1981). This antibody does not cross-react with pig, dog, mouse, or rat renin (Yokosawa et al. 1978). The antibody has been previously used to detect renin in normal human kidney (Faraggiana et al. 1982a) and in autosomal dominant polycystic kidney tissue (Torres et al. 1992; Loghman-Adham et al. 2004). Rabbit polyclonal anti-human AGT antibody was a generous gift of Dr Duane Tewksbury, Marshfield Medical Research Foundation (Marshfield, WI). It was used as antiserum without affinity purification. Rabbit polyclonal antibody (
Information on antibodies used
Results
Hematoxylin and eosin or periodic acid–Schiff stains of the ARPKD kidneys showed multiple dilated cystic tubules and large cysts extending from the cortex into the deep medulla, accompanied with loss of corticomedullary differentiation (not shown). Many glomeruli were visible between the more superficial cysts. No appreciable interstitial fibrosis could be seen in these neonatal kidney specimens.
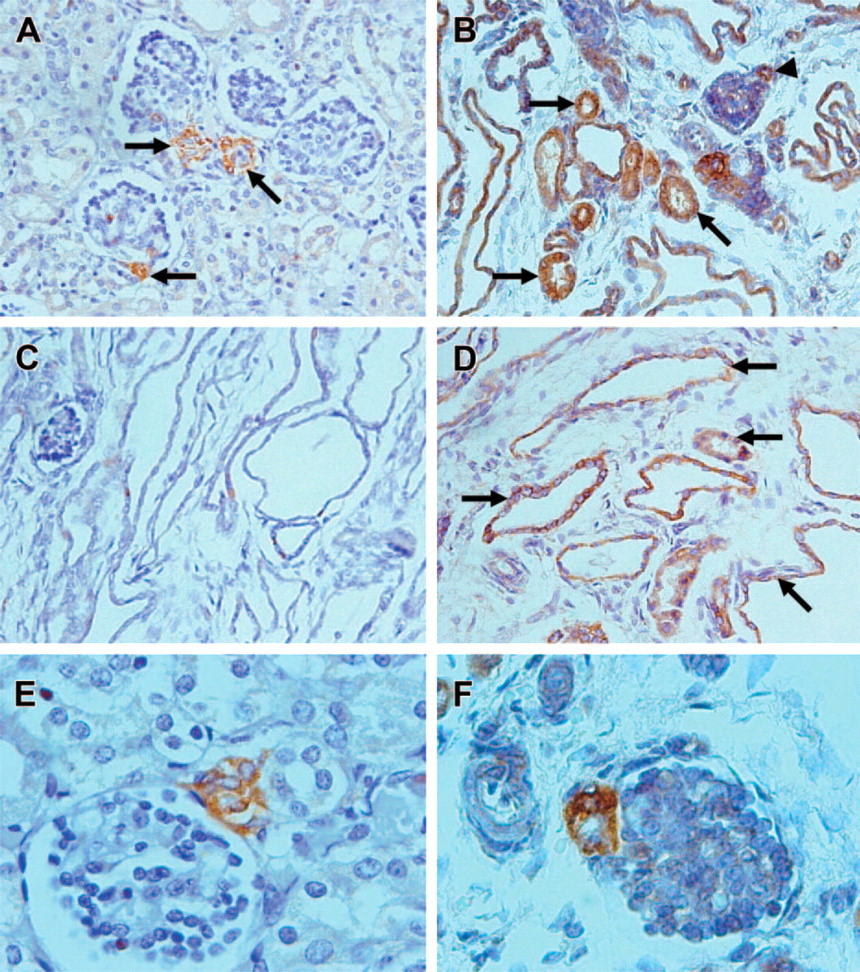
Previous studies have shown that in ARPKD kidneys, cysts are derived primarily from the collecting ducts (Verani et al. 1989). Proximal tubule-derived cysts have been observed in human ARPKD fetal kidney specimens, ranging in gestational age from 14 to 26 weeks (Nakanishi et al. 2000). To ascertain the tubule origin of the cysts in our infantile ARPKD kidney specimens, we used staining with lectins specific for proximal and distal tubules (Faraggiana et al. 1982b). LTA lectin was used as a marker of proximal tubules and PNA was used as a marker of distal/collecting tubules (Faraggiana et al. 1982b). In normal control kidneys, using the LTA lectin, we observed distinct staining of the brush borders of proximal tubules (Figure 1A). In ARPKD kidneys, we observed LTA staining of proximal tubules, which were larger and with less distinct brush borders than those of the normal kidney (Figure 1B). The distal tubules and the surrounding cysts did not bind LTA. Using PNA lectin, in normal control kidney sections, we observed extensive staining of distal tubules (Figure 1C). In ARPKD kidneys, we further showed PNA staining of many tubules and a large majority of the cysts, identifying their origin as distal tubule/collecting duct (Figure 1D). Occasional cysts did not stain with either LTA or PNA, suggesting they either originated from other nephron segments or had lost their ability to bind lectins.
Demonstration of tubule origin of cysts in autosomal recessive polycystic kidney disease (ARPKD) kidney. (
Patterns of renin expression in autosomal recessive polycystic kidney disease (ARPKD) kidney. (
Renin Expression in ARPKD Kidneys
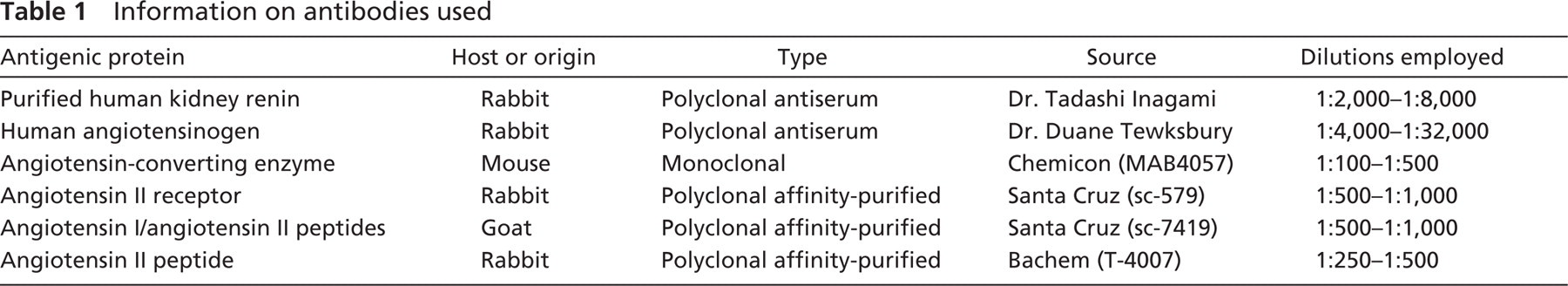
In control human kidneys, we observed renin staining confined to the afferent arterioles of some glomeruli visible at the highest antibody dilution used; there was no staining of tubules (Figure 2A). In ARPKD kidneys, we also observed renin staining in afferent arterioles of a few glomeruli (Figures 2B and 2F). However, a major difference with normal kidney was the presence of intense renin staining in many tubules (Figure 2B) and in the majority of the cysts (Figure 2D). In contrast to normal kidneys in which renin staining was seen primarily in glomeruli from superficial cortex, in ARPKD kidneys, renin-positive areas were mainly localized within the deep cortex and the medulla. When viewed at high magnification, renin staining was confined to tubular epithelial cells and to cyst-lining epithelia, except in afferent arterioles. No staining was observed when preimmune serum was used instead of the primary antibody (Figure 2C).
To determine the nephron origin of tubules that express renin, we studied their staining characteristics with specific lectins as described previously (Figure 3). Staining of adjacent sections of the same ARPKD kidney with renin antibody and with PNA and LTA lectins revealed that renin is localized primarily to cystlining cells and to distal tubules, identified by their ability to bind PNA lectin. Renin staining was also seen in some proximal tubules, identified by their ability to bind LTA but not PNA (Figure 3 A-C).
AGT Expression in ARPKD Kidneys
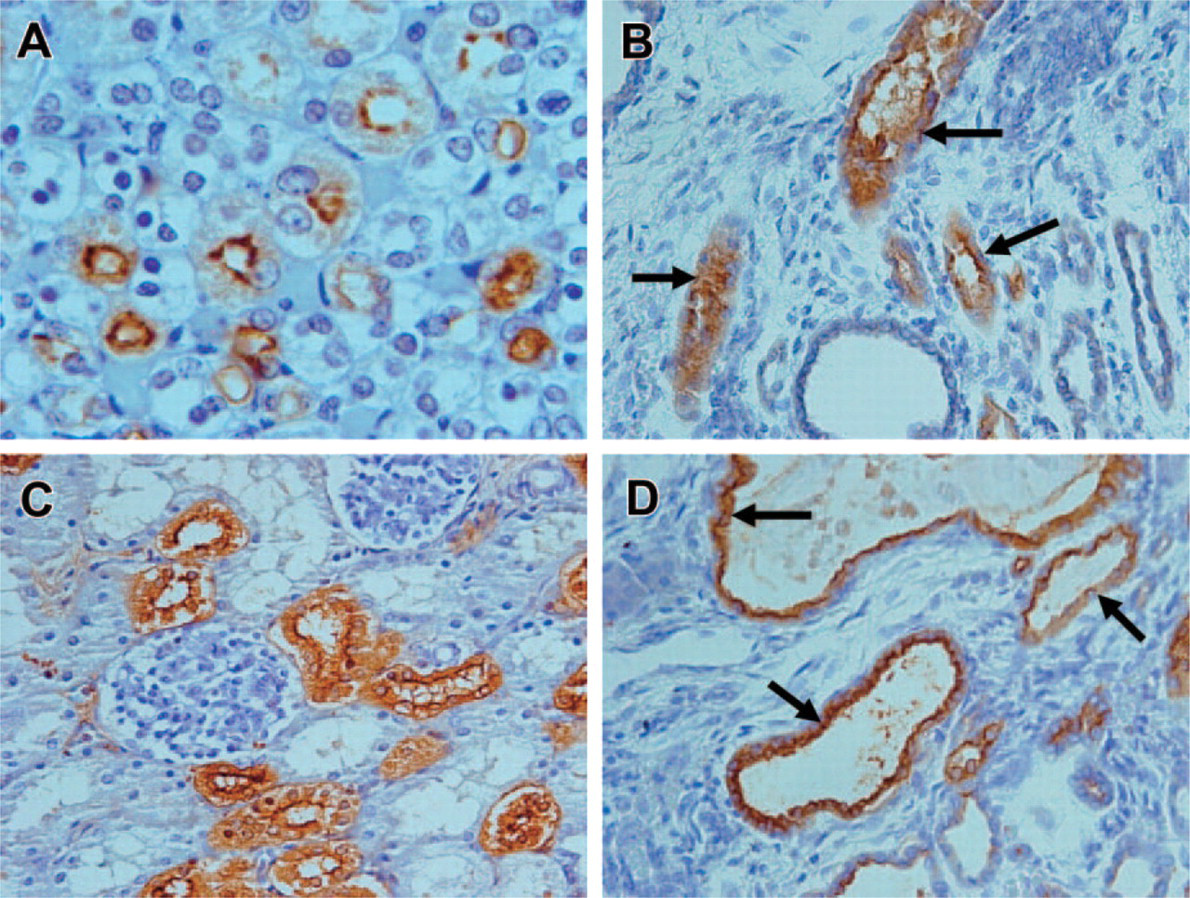
As previously reported by others, in normal control kidneys, AGT staining was confined to proximal tubules (Figures 4A and 4B). In ARPKD kidneys, intense AGT staining was seen in proximal tubules (Figures 4C and 4D). AGT staining was also seen in ∼15% of the cysts (Figure 4E). AGT staining of proximal tubules persisted with further antibody dilution down to 1:32,000, whereas AGT staining of cysts disappeared at antibody dilutions beyond 1:16,000. When compared with the staining pattern for renin (Figure 2), the number of cysts showing AGT staining was much lower than the number of cysts showing renin staining. No staining was observed when preimmune serum was used instead of the primary antibody (Figure 4F).
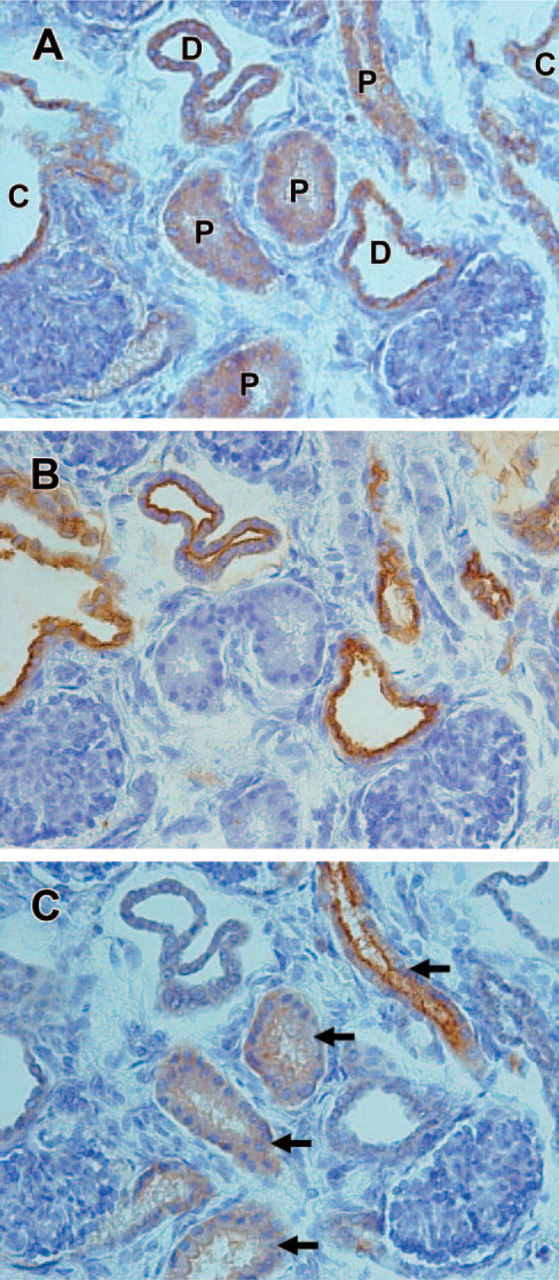
Determination of the origin of renin in autosomal recessive polycystic kidney disease (ARPKD) kidney. Serial sections of the same region of an ARPKD kidney containing tubules and two glomeruli were incubated with anti-renin antibody (
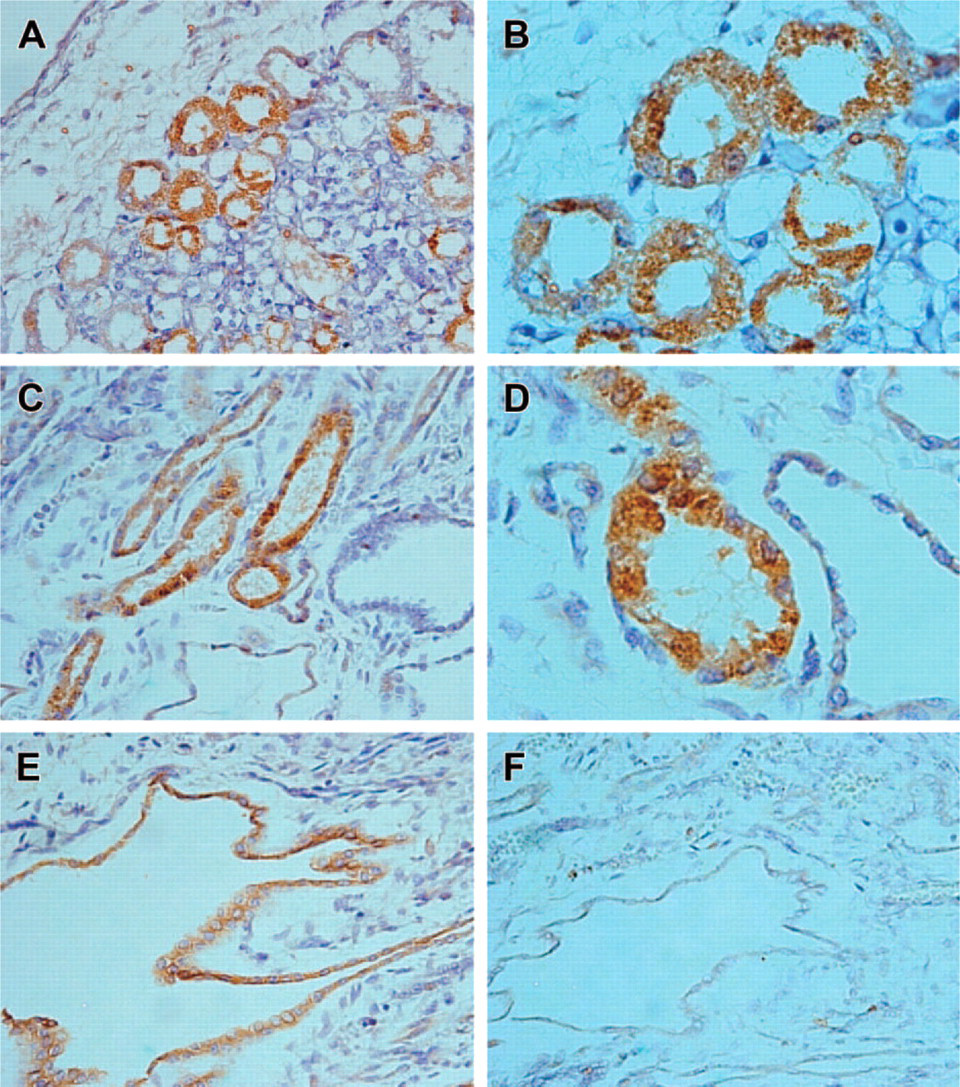
Angiotensinogen (AGT) expression in autosomal recessive polycystic kidney disease (ARPKD) kidney. (
Expression of Other RAS Components in ARPKD Kidneys
To determine if other components of the RAS are expressed by ARPKD kidneys, additional kidney sections were incubated with antibodies against ACE, Ang I and Ang II peptides, and AT1 receptor (Figure 5). In ARPKD kidneys, we observed ACE staining in many tubules and in ∼30% of the cysts (Figures 5B and 5C). The staining intensity of the tubules was higher in ARPKD kidney than in normal control kidney (compare Figures 5A and 5B). We also found strong staining for AT1 receptors in many tubules and in the majority of the cysts (Figures 5E and 5F). As reported previously in the normal kidney, AT1 receptor staining was also seen in the afferent arterioles of some glomeruli (not shown). Using an antibody that recognizes both Ang I and Ang II, we showed strong staining of many tubules and cysts (Figures 5G and 5H). Approximately half of the cysts showed Ang I/II staining. The expression of Ang II was confirmed using another antibody with high specificity for Ang II. No staining was observed when preimmune serum was used instead of the primary antibodies (Figure 5I). Taken together, the results indicate that all the components of the RAS are expressed in tubules and in cysts of ARPKD kidney.
Discussion
This is the first report of expression of components of the RAS in human ARPKD kidneys. The finding is remarkable by the extensive and ectopic nature of renin staining detected in these kidneys, which is far beyond what has been observed in other types of kidney diseases associated with hypertension (Taugner et al. 1982) or in end-stage renal disease (Faraggiana et al. 1988). Within the kidney, renin is produced by the afferent arterioles at the juxtaglomerular apparatus (JGA), whereas AGT is produced almost exclusively by the proximal tubules. Renin mRNA expression outside the JGA has been found in microdissected proximal tubules of rats, using the sensitive RT-PCR technique after stimulation of renin production by ACE inhibition (Moe et al. 1993). Renin expression has also been shown by immunohistochemistry in connecting tubules of mice placed on a sodium-restricted diet (Rohrwasser et al. 1999) and in both cortical and medullary collecting tubules of rats (Prieto-Carrasquero et al. 2004). Tubular renin and Ang II expression have also been reported in the remnant kidney of subtotally nephrectomized rats (Gilbert et al. 1999). Whether ectopic renin expression in tubules and cysts is merely a reflection of reduced nephron mass cannot be excluded, but this seems unlikely because of the more extensive nature of renin staining seen in ARPKD kidneys compared with remnant kidneys. Ectopic renin expression by cysts in ARPKD kidneys is not the result of the young age of the infants studied. Kidney sections from two infants without ARPKD did not show any renin staining outside of the JGA.
Recently, renin mRNA and protein expression were described in developing tubules and ureteric bud branches in avascular metanephric organ cultures (Norwood et al. 2000). Ureteric bud is the precursor of collecting tubules, which are dilated in ARPKD, suggesting that distal nephron renin expression may be derepressed in ARPKD. Accordingly, in ARPKD kidneys, renin was expressed almost exclusively by the distal tubules and cysts, identified by PNA lectin staining. Only occasional proximal tubules showed renin staining (Figure 2 and Figure 3). Because in ARPKD, cysts are derived from the collecting ducts (Verani et al. 1989; Silva et al. 1993), renin expression by cysts expands on previous reports that renin can be produced by the distal nephron (Rohrwasser et al. 1999; Prieto-Carrasquero et al. 2004). AGT was expressed in proximal tubules and in ∼15% of the cysts. To our knowledge, this is the first observation of AGT expression by cells originating from distal tubules or collecting ducts.
It could be argued that renin staining of tubules is a nonspecific finding and merely represents filtered renin endocytosed from the tubule lumen. This would be unlikely because, in ARPKD, the cysts originate from the collecting ducts. Filtered renin is degraded in the proximal tubule and ∼2% may reach the distal tubules and excreted in the urine (Kim et al. 1987). Because of its large molecular weight (∼57,000 kDa) (Tewksbury et al. 1978), AGT cannot be filtered by the glomerulus. However, AGT produced within the proximal tubules could reach the collecting ducts and the final urine (Rohrwasser et al. 1999). Therefore, we cannot exclude the possibility that AGT expression in cysts is a result of nonspecific uptake of AGT from the tubule lumen.
The use of archival kidney tissues to study the presence and distribution of components of the RAS provides limited information. Fresh tissue from nephrectomy specimens is difficult to obtain because of the rare occurrence of ARPKD. If available, additional studies such as mRNA analysis and more quantitative protein analysis by Western blotting could be performed. An immortalized cell line derived from ARPKD kidneys was recently described (Rohatgi et al. 2003). Attempts at demonstrating renin staining in these cells were unsuccessful. Additional studies, perhaps using a different antibody are warranted.
In a recent study of ADPKD kidneys, we showed ectopic renin and AGT expression by cyst epithelium (Loghman-Adham et al. 2004). Contrary to the findings reported here, in ADPKD kidneys, renin staining was not observed in afferent arterioles. We speculate that JGA renin may be downregulated in ADPKD. An alternative hypothesis is that many glomeruli were destroyed in these end-stage kidneys, leading to involution of their arterioles (Loghman-Adham et al. 2004). In contrast, ARPKD kidneys studied here were from young infants and had relatively well-preserved glomeruli. The expression of RAS components in cysts and tubules in two models of polycystic kidney disease suggests a common mechanism of hypertension in different forms of polycystic kidney disease.
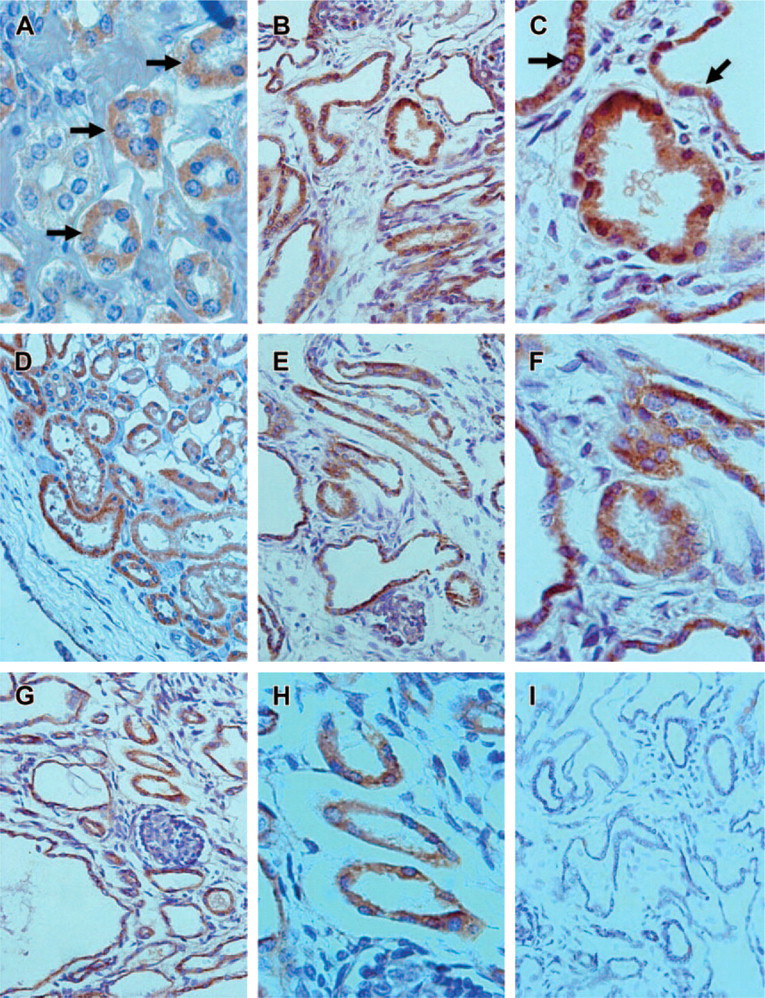
Demonstration of other components of the renin-angiotensin system in autosomal recessive polycystic kidney disease (ARPKD) kidney. (
This study provides strong evidence in support of the existence of a paracrine/autocrine intrarenal RAS in ARPKD kidneys (Figure 6). Renin and AGT production by tubules and cysts along with the presence of ACE at the same locations could result in the formation of Ang I and Ang II. Immunoreactive Ang I and Ang II is seen in tubules and cysts and is likely secreted into the lumen. Ang II could bind to specific receptors along the functioning tubules (Harrison-Bernard et al. 1997) and lead to increased salt and water retention via increased tubular sodium reabsorption both by the proximal tubules (Geibel et al. 1990) and by the collecting ducts (Wang and Giebisch 1996; Peti-Peterdi et al. 2002). Continued increased tubular sodium reabsorption may lead to a shift of the pressurenatriuresis curve to the right such that higher blood pressures would be needed to maintain sodium excretion (Hall et al. 1996,1999). The end result would be chronic hypertension, unless sodium intake is reduced. In support of this hypothesis is a recent study in immortalized cyst-lining epithelial cells from ARPKD kidneys that shows that these cells absorb sodium by an amiloride-sensitive pathway (Rohatgi et al. 2003). Another study, using collecting duct principal cells from a mouse model of ARPKD, suggests decreased amiloride-sensitive sodium absorption (Veizis et al. 2004). Despite these discrepancies observed in vitro, the net in vivo effect of Ang II excess appears to be increased sodium reabsorption perhaps due to a predominance of the absorptive compared with the secretory sodium fluxes in the functioning tubules.
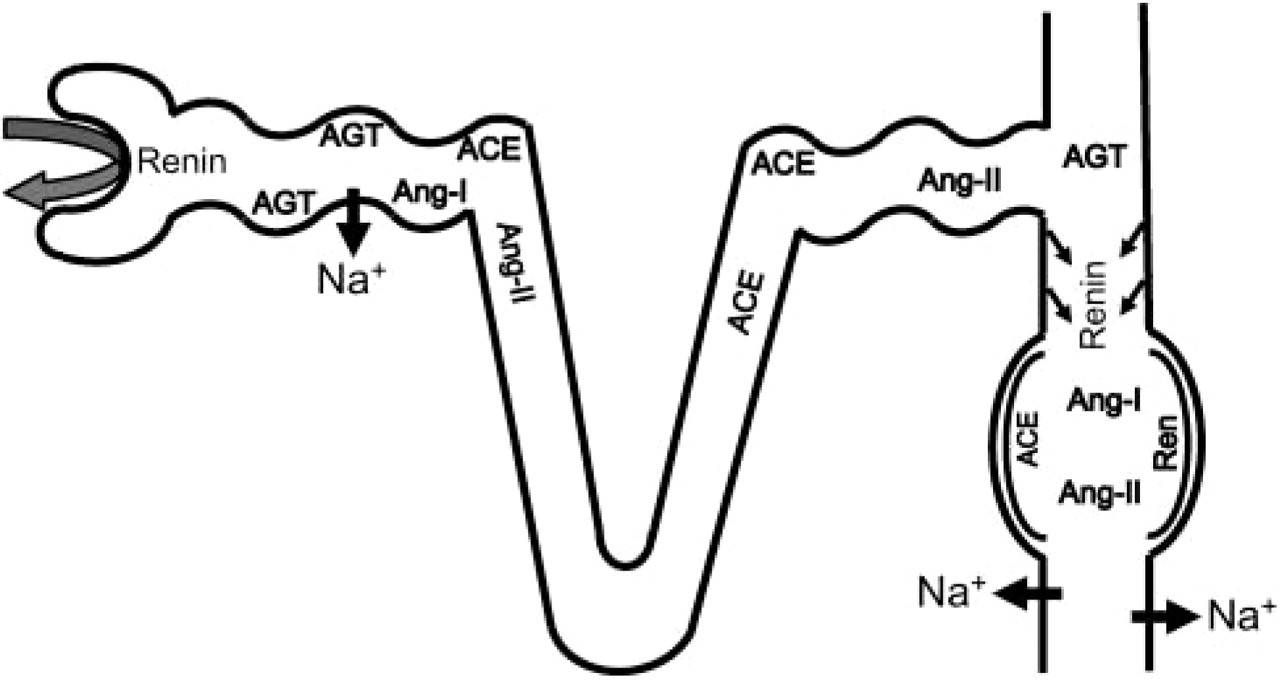
Schematic diagram of a nephron in autosomal recessive polycystic kidney disease (ARPKD) kidney showing the location of components of the renin-angiotensin system. Angiotensinogen (AGT) is produced by proximal tubules and can be cleaved by the filtered renin to form angiotensin I (Ang I), which is readily converted to angiotensin II (Ang II) by locally abundant angiotensin-converting enzyme. AGT can also remain in the tubule lumen and reach the distal nephron. Abundant renin is produced in the distal nephron, which includes distal tubules and cysts of collecting duct origin. Renin from the distal nephron sites and cysts can cleave the intraluminal AGT, resulting in the production of Ang I, followed by the formation of Ang II at these sites. The final result is increased Ang II concentrations, both within the proximal and distal tubules. Angiotensin II could stimulate sodium and water reabsorption at both nephron sites. The net result is salt and water retention, which may lead to hypertension, if salt intake is not reduced.
The reasons for ectopic expression of renin and other RAS components by cysts cannot be determined by the present studies. Mutations in a recently discovered gene, named PKHD1, result in ARPKD (Ward et al. 2002). PKHD1 encodes a 447-kDa, membrane-associated protein termed fibrocystin, which is localized to collecting tubules in normal kidneys, but absent from ARPKD kidneys (Harris and Rossetti 2004). The subcellular localization of fibrocystin to primary cilia is very similar to that described for polycystin 1 and 2 in ADPKD (Ward et al. 2003). This suggests that ciliary dysfunction may be linked to the pathogenesis of ARPKD.
Reduced intracellular Ca2+ concentration is known to stimulate renin production by the JGA cells (Schnermann 1998). Renin release from JGA cells is also controlled by changes in early distal flow rate. Increasing the flow rate depresses renin release, whereas reducing flow rate increases renin release (Leyssac 1986). Bending of cilia by mechanical flow initiates Ca2+ transients resulting in increased intracellular Ca2+ concentrations. Polycystins act both as chemosensors and as flow sensors in renal tubules and transduce mechanical fluid flow signals into Ca2+ signals (Nauli et al. 2003; Zhang et al. 2004). Nauli et al. (2003) have recently shown that the cilia in epithelial cells with a homozygous polycystin mutation fail to sense fluid flow. One could hypothesize that increased renin expression by cyst-lining epithelia in ARPKD is related to inability of the primary cilia to detect and transduce changes in flow or in the composition of the tubular fluid.
Footnotes
Acknowledgements
These studies were supported by grants from the Polycystic Kidney Research Foundation and by a Fleur-de-Lis grant from SSM-Cardinal Glennon Children's Hospital Foundation (to MLA) and Grant HL-58205 from National Institutes of Health (to TI). We thank Dr. Patricia Wilson, Mount Sinai School of Medicine, for studies in cyst-derived cells and Dr. Luis Salinas-Madrigal, St Louis University, for helpful discussions.